Gillian Ferko and Katie Swade
Summary of:
Ueno N, Hasebe T, Kaneko A, Yamamoto M, Fujiya M, Kohgo Y, Kono T, Wang CZ, Yuan CS, Bissonnette M, Chang EB, Musch MW. TU-100 (Daikenchuto) and ginger ameliorate anti-CD3 antibody induced T cell-mediated murine enteritis: microbe-independent effects involving Akt and NF-κB suppression. PLoS One. 2014 May 23;9(5):e97456. doi: 10.1371/journal.pone.0097456.
Background
What is Enteritis?
Enteritis, more commonly known as Crohn’s disease, is a chronic inflammatory disorder of the digestive or GI tract that affects 1.6 million Americans. Crohn’s affects people of all ages, but the exact cause of the disease is unknown. It can affect anyone, but is usually diagnosed between ages of 15 and 35 for males and females equally. Possible causes could be genetic or environmental factors (antigens in the environment that can stimulate inflammation). Symptoms include frequent diarrhea, abdominal pain and cramping, rectal bleeding, fatigue, weight loss, reduced appetite, and fever. There are periods of high intense symptoms, but the disease does not persist constantly. Up to 20% of the people with Crohn’s have a blood relative with IBD (Inflammatory Bowel Disorder)
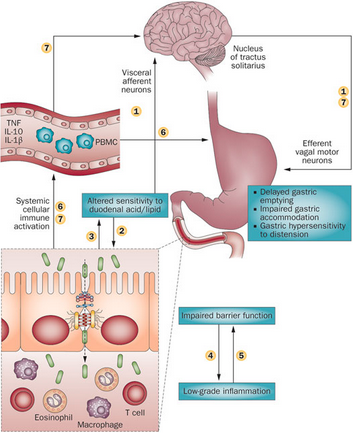
Figure 1. Schematic representation of the potential mechanism involved in the pathophysiology of Gastroenteritis.
Enteritis can be induced in the lab by treating cells or mice with anti-CD3 antibodies. These antibodies selectively activate T lymphocytes in the small intestine, which causes the pooling of intestinal contents, and eventually epithelial cell apoptosis. These antibodies have also been shown to increase TNF-⍺ levels in the small intestine and therefore are essential in the development of enteritis. This anti-CD3 antibody response was first discovered in humans in an attempt to suppress organ transplant rejection. Post-treatment, these patients developed a systemic cytokine response, indicating that this antibody leads to inflammation in the small intestine.
Japanese Traditional Medicine Background:
A traditional Japanese medicine, TU-100 (daikenchuto) is thought to have anti-inflammatory, pro-kinetic, and blood flow effects in the GI tract. The three main components of TU-100 are ginseng, ginger, and Japanese green pepper so the complex is valued as an all-natural, environmental, and traditional form of medicine. Ginger has anti-inflammatory and blood flow effects that can potentially modulate mitogen activated protein kinase, protein kinase B (Akt), and NF-kB activities.
Question: Can TU-100 or its components block the effects of Anti-CD3 antibody-induced enteritis?
Approach:
In all mouse studies, only germ-free (GF) and specific-pathogen free (SPF) mice were used to ensure the findings were independent of gut microbes. Mice were given TU-100 or ginger alone by diet or gavage for up to 24 hours then treated with anti-CD3 antibodies. The mice were then sacrificed and a laparotomy was performed to remove the intestine. A segment of the intestine was removed, weighed, and stained. RNA was extracted from this segment and analyzed for TNF-⍺ using Western blot and ELISA.
Additionally, human colonic adenocarcinoma (Caco2BBE) cells were treated with IFNy (100 U/ml) to increase TNF-⍺ receptor expression. These cells were co-cultured and treated with TU-100 with Human Jurkat cells to study the interaction and signaling between T-lymphocytes and epithelial cells.
Main Findings:
-Anti-CD3 antibodies were shown to cause a type of enteritis that is dependent on T cells and regulated by lymphocytes. This enteritis can be classified as gut microbe-independent and independent of intestinal bacteria.
-TU-100 and ginger alone, but not ginseng or Japanese pepper alone, blocked the anti-CD3 antibody-induced enteritis by significantly decreasing the amount of fluid accumulation within the jejunum and the stimulated expression of TNF-⍺.
-TU-100 also decreased the resulting apoptosis from anti-CD3 antibody treatment in jejunal epithelial cells.
-TU-100 and ginger were shown to block the activation of Akt after anti-CD3 antibody treatment, consistent with findings that ginger has been reported to block Akt activation.
-In the T-lymphocyte and epithelial cell co-culture, TU-100 and ginger show a block in anti-CD3 antibody induced activation of T-lymphocyte Akt and subsequent TNF-⍺ activation of epithelial NFkB.
-These findings demonstrate that TU-100 and ginger do in fact block the effects of anti-CD3 antibody induced enteritis.
Broader Context:
Enteritis is classified as a Inflammatory Bowel Disorder (IBD), along with a disorder called Ulcerative Colitis. Enteritis is associated with inflammation in any part of the the GI tract, while Ulcerative Colitis is associated with inflammation only in the colon or large intestine. These disorders, along with other small bowel inflammatory diseases like viral enteritis or Ciliac’s disease, remain unclear in regards to cure or effective treatment. Understanding the pathophysiology of enteritis and the effects of TU-100, ginger, or other substances can help extend the potential therapies for gastrointestinal disorders.
References:
https://www.crohnsandcolitis.com/crohns