Mike Aimino & Lisa Freeman
Summary of:
Clement, L.C., Avila-Casado, C., Macé, C., Soria, E., Bakker, W.W., Kersten, S. & Chugh, S.S. (2011). Podocyte-secreted angiopoietin-like-4 mediates proteinuria in glucocorticoid-sensitive nephrotic syndrome. Nature Medicine, 17(1), 117-122. doi: 10.1038/nm.2261.
What is Nephrotic Syndrome?
Nephrotic syndrome is a condition that causes proteinuria, hypoalbuminemia, edema, hyperlipidemia, and lipiduria in individuals that have it. Proetinuria and lipiduria are when there are proteins and lipids in the urine, respectively. Hypoalbuminemia is when there is a low amount of albumin in the blood while hyperlipidemia is when there are high amounts of lipids in the blood. It can be caused by diabetic nephropathy, minimal change disease (MCD), focal and segmental glomerulosclerosis (FSGS), or membranous nephropathy. MCD is the primary cause of nephrotic syndrome in pre-adolescents, making up 85-95% of the cases. About 15 in 100,000 children have MCD with 2-7 new cases annually in 100,000 children. The prevalence of MCD is much lower in adults, making up only 10-15% of the cases. MCD is sensitive to glucocorticoid treatment, while the other diseases show a varied response, making it a good target to study.
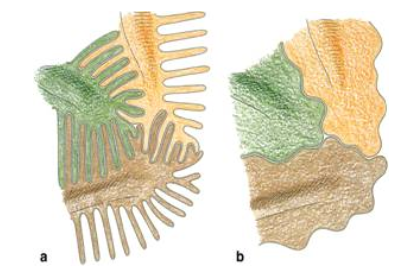
Other complications of nephrotic syndrome include foot process effacement of podocytes. Normally, podocytes extend primary processes to the glomerular basement membrane (GBM) of the capillaries. Foot processes extend from the primary processes and lie on the GBM. Adjacent foot processes then interdigitate, which looks similar to locking your fingers together (pictured below). In individuals with necrotic syndrome, the foot processes disappear, making it look like the cell membrane is continuous. This leaves spaces between podocytes which allows proteins to leave and enter the urine.
All angioproietin-like proteins (Angptl) are glycoproteins that are sensitive to glucocorticoids. Angiopoietin-like proteins have been found to play a role in the development of hypertriglyceridemia and tumor metastasis. They have many different effects on cells depending on the part of the body in which they are found. Angptl4 is an inhibitor of lipoprotein lipase and has an effect on triglyceride levels in the blood. There has been no previous research to show that Angptl4 plays a role in proteinuria.
Question
What role, if any, does Angptl4 play in proteinuria associated with nephrotic syndrome?
Approach
The researchers employed a large arsenal of experiments and analysis techniques, so we will only give an overview of the experimental approach here. They started by evaluating four different nephropathy models, each model simulating a different disease that causes nephrotic syndrome. They identified the model that yielded the greatest increase in Angptl4 expression (puromycin nephrosis, or PAN), and used this model in later experimentation. They then studied a previously established Angptl4 transgenic mouse model and developed two new transgenic rat models. The NPHS2-Angptl4 model is characterized by upregulated Angptl4 in podocytes, while the aP2-Angptl4 is characterized by an upregulation of circulating Angptl4, secreted from adipose tissue. They evaluated Angptl4 expression and morphological changes for each model.
Although data for the aP2-Angptl4 is presented in supplemental materials, the remainder of the paper is focused on continuing experimentation with the NPHS2-Angptl4 model. With this model, they measured albuminuria in rats at varying ages. They then induced PAN, a model for minimal change disease (MCD), and measured albuminuria again. To analyze an additional variable affecting protein expression, they treated the rats with glucocorticoids after inducing PAN and measured resulting proteinuria and Angptl4 expression. Finally, the researchers conducted an in vitro study in two different cell lines of the sialylation of Angptl4 on its electrophoretic migration and the level of proteinuria occurring in the transgenic models.
A variety of analysis techniques were utilized throughout the experiments described above. Light microscopy and electron microscopy were used to analyze morphological changes at the glomerular and cellular levels, respectively. Immunohistochemistry with confocal microscopy and immunogold electron microscopy were used to localize and quantify Angptl4 expression. The researchers used SDS-PAGE to detect urinary protein and a combination of 2-D gel electrophoresis and western blotting to differentiate between forms of Angptl4.
Main Findings
A key result from these experiments was that increased expression of podocyte-secreted Angptl4 (NPHS2-Angptl4 transgenic model) caused increased proteinuria, similarly to proteinuria observed in rat models and human MCD patients. Angptl4 expression was also associated with morphological changes characteristic of MCD. More specifically, proteinuria was induced when the glomerular basement membrane (GBM) showed the presence of Angptl4, even though the podocytes did not yet show morphological changes. The researchers interpreted these results as an indication that Angptl4 causes a defect in the GBM that ultimately leads to proteinuria. They also found that Angptl4 decreased after glucocorticoid treatment. Finally, their results showed that sialylation of Angptl4 was associated with decreased proteinuria.
Broader Context
The results show that in the PAN model there is a 60-80 fold upregulation of glomerular Angptl4 expression in the PAN model. This is close to the 120 fold increase found in the NPHS2-Angptl4 heterozygous, male rats, meaning that it is a good model for studying nephrotic syndrome. This model is the first demonstration of the important role Angptl4 plays in proteinuria. Because the NPHS2-Angptl4 rats had reduced albuminuria when fed with ManNac and had an increase in the sialylation of glomerular Angptl4, it suggests that hyposialylation could be a mechanism by which Anglt4 overexpression causes proteinuria. Therefore, treatment with sialic acid precursors could be a potential therapy for individuals with some forms of nephrotic syndrome, particularly minimal change disease.
References
2. National Institute of Diabetes and Digestive and Kidney Diseases. Glomerular Disease Primer: The Normal Kidney. http://www.niddk.nih.gov/research-funding/at-niddk/labs-branches/kidney-disease-branch/kidney-diseases-section/glomerular-disease-primer/-normal-kidney/Pages/normal-kidneys.aspx