Heather Geist and Juliana Schneider
Summary of:
Wee, CD, Havens, MA, Jodelka, FM, Hastings, ML. (2014). Targeting SR Proteins Improves SMN Expression in Spinal Muscular Atrophy Cells. PLoS ONE, 9(12). e115205. doi:10.1371/journal.pone.0115205
Background:
What is SMA?
Spinal muscular atrophy (SMA) is an autosomal recessive genetic disease that is one of the most common inherited causes of pediatric mortality. SMA has occurs in 1 in 6000 live births and has a carrier frequency of 1 in 40. This disease affects the voluntary muscle movement controls of the nervous system. SMA is categorized as a degenerative neuromuscular disease, which involves the loss of motor neurons in the spinal cord. This in turn prevents the muscle from receiving signals, leading to muscular atrophy over time. The most common form of SMA is called chromosome 5 SMA, which has great variability in age of onset. For this reason it is typically classified into types 1 through 4. Our main focus will be on Type I SMA patients. Type I is correlated with the lowest level of functioning patients. Onset of symptoms in Type I patients usually occurs at birth or during infancy, leading to the greatest impact on motor function. The most drastically affected muscles are typically those closest to the center of the body and because of early onset, these individuals have high mortality rates.
What is SMA Caused by?
SMA is caused by deletions or mutations in the Survival of Motor Neuron I gene (SMN1) located on chromosome 5. SMN proteins are essential for conventional motor neuron function. If a deletion or mutation of SMN1 occurs, an unstable form of the SMN protein results. The SMN2 gene, also located on chromosome 5, has the ability to produce SMN protein at smaller volumes (Figure 1). The main aberration between SMN1 and 2 is the single nucleotide in exon7, which is thought to be an exon splice enhancer. As a result of this single nucleotide difference, SMN2 cannot compensate for the loss of SMN1. The SMN1 gene is telomeric. It contains more than 4 genes, so it is susceptible to rearrangements and deletions. SMN2, however, is centromeric and is not as prone to rearrangement. Interestingly, increasing the inclusion of exon7 escalates the abundance of SMN protein. The SMN2 gene, also located on chromosome 5, produces SMN protein at smaller volumes. Inclusion of exon7 has been shown to have efficacy in animal models of SMA and early human clinical trials.
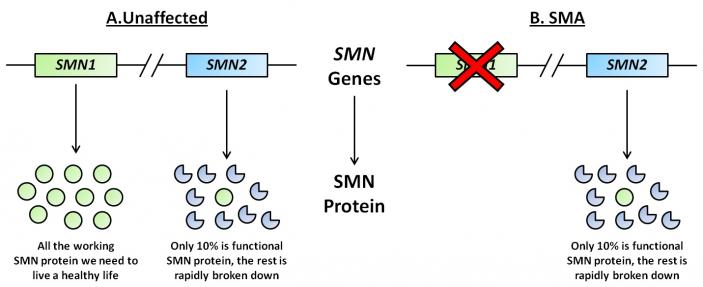 Figure 1. Representative diagram of SMN1 and SMN2 genes and subsequent proteins in unaffected and SMA individuals. (Image modified from http://www.smasupportuk.org.uk/blog/ research/azzouz-laboratory-gene-therapy-for-sma).
Figure 1. Representative diagram of SMN1 and SMN2 genes and subsequent proteins in unaffected and SMA individuals. (Image modified from http://www.smasupportuk.org.uk/blog/ research/azzouz-laboratory-gene-therapy-for-sma).
Splicing and its role in SMA:
Exons and introns undergo a period of pre-mRNA splicing. This is conducted by the spliceosome complex, which is comprised of 5 snRNPs and other splicing proteins/factors (Figure 2). The splicesome complex works to identify exons and introns through binding to specific consensus splicing sequences on both the 5’ and 3’ ends of mRNA. Multiple proteins and sequence elements have been shown to play a role in regulating the splicing of SMN exon7. Specifically, SRSF1, SRSF2, and SRSF9 are known to influence exon7 inclusion, but few other members of the SR protein family have been explored regarding their role in SMN2 exon7 splicing. In addition to the SR protein family, the hnRNP proteins have also been implicated in play a role in exon 7 inclusion or exclusion.
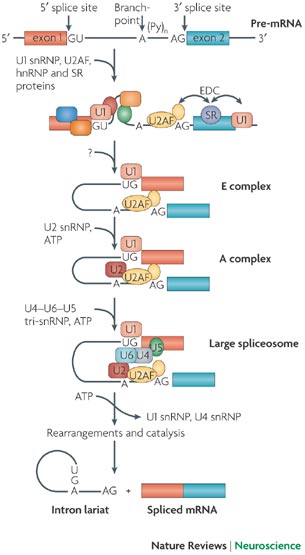 Figure 2. The spliceosome complex and process of pre-mRNA processing. (Image modified from http://www.nature.com/nrn/journal/v8/n11/box/nrn2237_BX1.html)
Figure 2. The spliceosome complex and process of pre-mRNA processing. (Image modified from http://www.nature.com/nrn/journal/v8/n11/box/nrn2237_BX1.html)
Question:
What characteristics and roles do SR and hnRNP proteins play in SMA disease pathogenesis? Do any of these proteins have therapeutic implications?
Experimental Approach:
Two main cell-lines were utilized throughout the experiments. The first was HeLa cells, which are derived from a cervical cancer tumor. The cells were treated and transfected with siRNA and Lipofectamine to knockdown specific SR and hnRNPs. In order to overexpress these proteins, expression vectors were transfected into the cells. The second cell-line used were human fibroblast cells derived from a Type I SMA patient only having one copy of SMN2. These cells were transfected as the HeLa cells were to knockdown the designated SR and hnRNP proteins. After cells were treated, RNA was isolated, and RT-PCR was conducted.
Proteins were extracted from the cells and immunoblots were performed by probing with 12 different antibodies directed at specific proteins. After quantitative analysis of fluorescence was performed, different levels of exon7 expression were observed. In vitro transcription was conducted on RNA splicing substrates with or without additional SR proteins. PCR analysis was done to quantify exon7 in these cells.
Key Results:
The most valuable result gathered from this experiment was the determined involvement of SRSF2 and SRSF3 in exon7 inclusion. Through knockdown of specific targets, an increased level of SMN protein was found to be present in these cells. This leads to further implications that these proteins could be therapeutic targets for Type I SMA patients.
Succeeding overexpression of SR proteins, the researchers observed a decrease in exon7 inclusion. Moreover, following successful knockdown of SR proteins in HeLa cells, it was found that exon7 inclusion increased significantly in 9 of 12 SR proteins. Specifically, exon7 inclusion was found to have the highest levels following SRSF3 knockdown. Following 40-50% knockdown of hnRNP A2B1 and U, a significant inclusion of exon7 resulted. This result confirmed that both SR and hnRNP proteins enhance splicing and through inhibiting their function, more exon 7 inclusion occurs.
Similar to the above experiments, the researchers decided to knockdown SR proteins in SMA cells and observe not only the effects on exon7 inclusion, but also SMN protein abundance. It was observed that upon knockdown of SRSF2 and 3, SMN protein levels increase. Thus, downregulation of these SR proteins leads to an increase in SMN protein levels. These results were consistent with the results obtained from HeLa cells. It was through this data that the researchers decided to examine the SRSF2 and 3 proteins more closely and deemed them to be the most effective therapeutic targets.
Broader Context:
There is a lower toxicity associated with targeting individual proteins such as splicing factors that have fundamental functions in the cell. Downregulation of single or multiple SR protein inhibitors of SMN2 exon7 inclusion could result in an improvement in full-length SMN protein. As a result, levels of SMN expression could increase to adequate therapeutic levels without fully disrupting other necessary functions of splicing factors.
Additional References:
MDA: Fighting Muscular Disease Website: mda.org/disease/spinal-muscular-atrophy/overview
Gene Cards: The Human Gene compendium Website: www.genecards.org
I find this article very interesting, and it seems like a crucial preliminary step in understanding Spinal Muscular Atrophy. One question that I have is how they chose to isolate the regulatory sequences around Exon 7. I am interested if other sequences would show the same results, and understanding more about how they chose to isolate this section.
The authors seem fairly confident in the potential therapeutic effects that SR knockdown, specifically that it would only take “modest down-regulation” for therapeutic efficacy. Their results show, in one case at least, about a 50% increase in SMN protein levels. Is there strong evidence that this amount of increase is sufficient to act therapeutically? Is their indication from previous research that this level of SMN protein would restore normal motor neuron function?
اینجا فارسی دانلود اهنگ
I’ll be honest, I usually ignore promotional offers on gaming platforms because they often feel too complicated or come with hidden conditions, but recently I decided to give Zoome Casino a try after seeing it mentioned in a discussion thread. What caught my attention wasn’t flashy advertising, but the idea that I could explore the platform without risking my own money right away. I activated the zoome casino no deposit bonus, and for the first time, I actually felt like I had the freedom to test different games at my own pace. I spent some time trying out slots and even a couple of table games, just to understand how everything worked. Later, I noticed additional incentives tied to my first deposit, which made me consider going further, but I appreciated that I didn’t feel pressured to do so immediately. The structure of these offers seemed more straightforward than what I’ve seen elsewhere, and that made a big difference in my overall impression. I’m still a casual player, but now I occasionally check back to see if there are any new promotions worth trying. Has anyone else found that these kinds of starting perks actually change how you approach a new platform, or do you still prefer to jump in without using them?
The authors seem fairly confident in the potential therapeutic effects that SR knockdown, specifically that it would only take “modest down-regulation” for therapeutic efficacy. Their results show, in one case at least, about a 50% increase in SMN protein levels. Is there strong evidence that this amount of increase is sufficient to act therapeutically? Is their indication from previous research that this level of SMN protein would restore normal motor neuron function?
As the authors noted in the second part of the results, overexpression of some SR proteins (e.g. SRSF4, 6, and 9) did not have a significant effect on exon 7 splicing. They rationalize these results by saying 1) they may not be involved in exon 7 splicing, 2) there was no significant change because their levels of overexpression were insufficient, or 3) endogenous proteins are already optimized for regulating splicing and cannot be further enhanced. What else could the authors do to verify these results and justify why there was no significant effect with these SR proteins?
The authors chose to analyze the sequences flanking exon 7 within 50 nucleotides but acknowledge that there are antisense oligonucleotides directed to sequences more distant from exon 7 which have been identified to improve exon 7 inclusion. I was wondering why they chose to only look at the flanking sequences. Is there any benefit in one over the other, or was it just a need to limit their analyses and was chosen arbitrarily? On that note I’m curious to see if there have been studies looking at those more distant or if the authors plan to analyze those options in the future as well.
In this study, the authors conclude that the majority of SR proteins (SRF1 and more) and two hnRNPS are regulators of SMN2 exon 7 inclusion. How is it that so many proteins are responsible for the exon 7 inclusion? Also,the study identified regulators of exon 7 inclusion and also confirmed the activity of proteins that have been tested for exon 7 inclusion in recent studies. However, SRF9 had no significant affect on SRF9. It would be interesting to verify the role of SRF9 and see why it may not affect exon 7 inclusion.
I am very intrigued by the research that the authors in this article present. There are not many therapeutics available for spinal muscular atrophy, and this disease often effects infants so an effective therapeutic has been sought out for awhile. Although the authors determined that the SRSF2 and 3 proteins increase SMN levels, I’m curious if they investigated other areas on exon 7 that contribute to protein production. I’m also curious to see how they implement these findings into a therapeutic for this disease.
I found this article very interesting, but I am still unclear as to how the authors propose their findings be translated into therapies for SMA. They state that downregulation of single or multiple SR protein inhibitors of SMN2 exon 7 inclusion would be most effective, but how would they determine which inhibitors to use? Is it enough to just inhibit SRSF 2 and 3 or would inhibition of most of the proteins involved in the complex be necessary and how would that distinction be made?
I found the aim of this paper very interesting. While we all have learned that pre-mRNA can be alternatively spliced based on tissue location or specific promoters present in a certain cell type, I had not considered alternative slicing as a therapeutic option for disease. Here, the idea is clear, two types of the gene exist, and one (SMN2) could potentially act as the main gene (SMN1) if it expressed exon 7. I would be interested to see this effects in an animal model. In addition I am very curious as to how this type of therapy could be applied to other human disease where a redundant gene is not present.
I think it is interesting that the splicing proteins are interacting in such a complicated way. As I believe the authors mentioned, wouldn’t knocking down one of the genes have an effect that is based at least partly on how that protein interacts with the other components of the spliceosome? With such complicated interactions, how much does this tell you about the function of one protein? Additionally, I wonder if the spliceosome operates the same way in the cell-free condition?
This paper provides some interesting results to consider, but I think the claims of therapeutic benefit are weak based on the data presented in this paper. The effects of altering single splicing proteins are modest at best and I wonder if these subtle differences would lead to any therapeutic benefit. I also have concern that the method they used to measure the transcript levels (semi-quantitative RT-pcr) was not the best way to do this. We can discuss semi-quantitative pcr in class on Friday, but it is not as quantitative (hence the word “semi”!) as the RT-pcr methods we discussed/are using in our lab.
I could understand the basic mechanism of SMA! I think figure 4 is strongest figure that can support the main idea of the paper that depleting inhibitor (SRSF2/SRSF3) would increase SMN2 exon 7 inclusion and SMN protein that can lead to an therapeutic agent. Thus, I really like the fact that this paper has clear outline, which tells splicing factors are important. However, I agree that this needs more detail on therapies of SMA. This only identified the fact that knocking down SRSF3 could increase in SMN protein, which is related to SMA. Instead of identifying the fact, I think it should go further from exon 7 inclusion. Exon 7 inclusion wouldn’t be the only reason that lead to SMA. Even in figure 5, I think it should go further with molecular pathway of SMA instead of confirming knocking down SRSF3 could be a potential therapeutic target. I was wondering why they were using semi-quantitative rt-pcr. I also was wondering why the data they got about SRSF9 was different than previously published data. The reason they stated was ambiguous (vague).
After reading both the journal article and the blog post I am still finding it difficult to understand the purpose and function of the different cell lines that were used and transfected with in the experimental approach? I also would like to further elaborate on this idea of two genes coding for same protein with the exception that SMN2 cannot account for loss of SMN1 but it increases the relative protein levels upon inclusion of this exon 7. What is it biochemically that allows for different functions of SMN1 and 2 based on the presence of Exon 7?
This article is absolutely remarkable, I’m so thankful for your share. Wishing you success! Free Valid AD0-E121 exam guide files test papers are here.
AACE International学習教材は、学習者が製品を使用するのに不便がないように役立つ複数の機能と思いやりのあるサービスを提供します。 CCP学習教材を購入し、しばらくの間辛抱強く学習すれば、わずかな失敗確率でCCPテストに合格することを保証できます。私たちの製品の価格はあなたが購入できる範囲内であり、私たちの学習教材を使用した後、あなたは確かに製品の価値があなたが支払う金額をはるかに超えていると感じるでしょう。 CCP学習ガイドを選択することは、Certified Cost Professional (CCP) Exam成功と完璧なサービスを選択することと同じです。
Your article is a work of art, thank you for sharing it with us! I’m sharing the C_SIGDA_2403 reliable exam topics exam materials for free—best of luck!
This article is fantastic, thank you for sharing this insight. Here are the free 1z0-1123-24 valid exam collection materials resources—good luck, everyone!
The content is too good not to like. Wish me success as I prepare for the Exam CISSP tutorials exam!
Your article is brilliant beyond words, I’m thankful for your share! Wishing everyone the best of luck! Free SPLK-1004 valid test experience questions are available!
The article was full of rich, valuable content. Free Reliable new IIA-CIA-Part1 test format exam papers—your path to promotion and salary increase!
Your article is truly awe-inspiring, thank you for sharing! I’m sharing the IIA-CHAL-QISA valid test cram sheet exam materials—best of luck to everyone!
I really appreciate your article, it was so insightful. This SPLK-1004 practical information helped me get promoted and receive a raise. Now I’m offering it for free. Wishing you all success in your career paths!
Wenn man an sich glaubt, kann man wirklich was erreichen. Der Grund, warum DeutschPrüfung jedem IT-Fachmann helfen kann, liegt in seiner Fähigkeit. Die Prüfungsmaterialien zur Huawei H31-311_V2.5 Zertifizierung von DeutschPrüfung können Ihnen zum Erfolg verhelfen. Jede Beschränkung fängt im Herzen an. Wenn Sie die Huawei H31-311_V2.5 Prüfung bestehen wollen, werden Sie DeutschPrüfung wählen. Eigentlich ist die Distanz zwischen Erfolg und Niederlage nicht weit. DeutschPrüfung führt Sie zum Erfolg.
EMC인증 D-PST-DY-23덤프로EMC시험을 패스,하지 못하셨다구요? 최선을 다했는데도 실패하였다는 말은 영원히 하지마세요. EMC인증 D-PST-DY-23시험을 패스하는 방법은 많고도 많습니다. ITDumpsKR의EMC인증 D-PST-DY-23덤프로 시험에 다시 도전해보세요. ITDumpsKR의EMC인증 D-PST-DY-23덤프는 착한 가격에 100%에 달하는 적중율과 패스율을 보장해드립니다. 시험에서 불합격성적표를 받으시면 덤프구매시 지불한 덤프비용을 환불처리해드립니다. ITDumpsKR의EMC인증 D-PST-DY-23덤프로 시험패스를 꿈꿔보세요.
This article is so uplifting, thank you for sharing it! Enhance your IT abilities and get free Latest GB0-392 exam discount voucher. Best wishes for success!
Your article really made an impression, thank you so much. Free Exam Salesforce-MuleSoft-Developer-II fees shared to help you improve your IT skills. Best of luck!
Itexamdump에서 판매하고 있는 Amazon AIF-C01인증시험자료는 시중에서 가장 최신버전으로서 시험적중율이 100%에 가깝습니다. Amazon AIF-C01덤프자료를 항상 최신버전으로 보장해드리기 위해Amazon AIF-C01시험문제가 변경되면 덤프자료를 업데이트하도록 최선을 다하고 있습니다. Itexamdump는 여러분이 자격증을 취득하는 길에서 없어서는 안되는 동반자로 되어드릴것을 약속해드립니다.
Your article was so insightful, I’ll carry it with me. I used the D-PDC-DY-23 test registration to get promoted and get a pay raise. Now it’s free to all. I hope you all get promoted soon!
Thank you for this amazing article, it left a profound impression on me. Wishing everyone the best of luck—free Valid ANS-C01 exam cram pdf materials available now!
This is such a motivational piece, thank you for sharing! This is the Valid 300-540 exam questions pdf exam I took for my promotion and salary increase. It’s free for you today—wishing you all success in your careers!
I’m deeply moved by your article, thank you for sharing it. Free Exam discount L4M6 voucher test questions—unlock promotions and salary growth today!
Thank you for sharing this inspiring article with us! Latest C-STC-2405 exam simulator free content is excellent, and you can access it completely for free.
Your article is incredibly inspiring, thank you for sharing! I was promoted and received a raise with the help of the Latest 156-587 exam cram. Now it’s available for free to all. Best wishes for your promotions!
ExamPassdump는 유일하게 여러분이 원하는SAP인증C_TS4FI_2023시험관련자료를 해결해드릴 수 잇는 사이트입니다. ExamPassdump에서 제공하는 자료로 응시는 문제없습니다, 여러분은 고득점으로 시험을 통과할 것입니다.
Reading this article, I have gained a completely new understanding of the issue. Free D-MN-OE-23 valid exam vce test questions—unlock promotions and salary growth today!
This article is mind-blowing, thank you for sharing! The D-PE-OE-23 certification exam infor test helped me secure my promotion and raise—grab it for free today!
Da qualche tempo mi dedico con maggiore attenzione al benessere personale e alla gestione della mia routine quotidiana. Cercando informazioni su prodotti specifici e articolari farmaceutici, ho scoperto diverse piattaforme specializzate. Tra quelle che ho consultato più spesso c’è farmacia italia shop , che mi ha colpito per la varietà delle categorie disponibili e per la facilità con cui è possibile trovare dettagli utili sui prodotti. Prima di effettuare qualsiasi scelta, mi piace confrontare caratteristiche, descrizioni e informazioni generali, e proprio per questo passo molto tempo a leggere contenuti aggiornati. farmacia italia shop è diventato uno dei riferimenti che tengo in considerazione quando desidero approfondire novità del settore e valutare diverse opzioni. Apprezzo soprattutto la struttura ordinata del catalogo e la possibilità di orientarmi rapidamente tra le varie sezioni. Per chi ama informarsi con calma e raccogliere dati prima di prendere decisioni, ritengo che farmacia italia shop rappresenti una risorsa interessante da esplorare.
I’m deeply impressed by your article, thank you for sharing it! The SPLK-2003 reliable exam bootcamp questions are essential for your promotion and salary increase—free now!
Thank you for the article, it really broadened my outlook. The Latest test LLQP collection materials resources are top-quality and free to use.
randm box 12k
ユーザーエクスペリエンスとクライアントのフィードバックを優先します。GWEB実践ガイドは、常にサービスを改善し、バージョンを更新してクライアントの利便性を高め、満足させるようにします。 GWEBトレーニング資料に関するクライアントの満足度は、前進を続ける原動力の源です。 GWEBガイド資料を理解できるようになりました。 GWEB認定に関する知識の主流の微妙な変更はすべてキャッチされ、利用可能なGWEB学習資料リソースの検索に最善を尽くします。
The viewpoints in the article have been very insightful, and I’ve learned a great deal. I used the Free NS0-516 practice test exam to get promoted and earn more. Now, it’s available for free. Best of luck with your career!
The content is on fire—like is clicked. We’ve made NS0-604 Pass4sure study materials available free of charge, hoping it enriches your studies.
Thank you for your article, it really captured my attention. Good luck on your exams! Here are the free C-THR89-2405 reliable practice exam online questions to help you out!
I deeply appreciate your share, this article is simply astonishing! Free 020-100 exam tutorial questions are shared. Good luck with your exam preparation!
我々の提供する資料は高質量で的中率も高いです。このC_THR85_2411模擬問題集を利用して、試験に参加するあなたはC_THR85_2411試験に合格できると信じています。ご安心に我々の問題集を利用してください。我々はあなたに最大の利便性をもたらすために、一番いいC_THR85_2411問題集を提供して、あなたが合格できるのを確保します。
The article is excellently written, and it has benefited me greatly. Sharing the Valid dumps AI-900 free materials for free—wishing you good luck!
インタネット時代に当たるなので、パソコン上のMicrosoftのPL-300試験についての情報は複雑で区別するのは困難なことであると思われます。それで、我々Topexamの高質で完備なPL-300問題集を勧めて、あなたの資料を選んでかかる時間のロースを減少し、もっと多くの時間を利用してPL-300問題集を勉強します。
他人の気付いていないときに、だんだんSAPのC-S4CPB-2408-JPN試験成功したいのですか?我が社はIT資格認証試験資料の販売者として、いつまでもできご客様に相応しく信頼できるC-S4CPB-2408-JPN問題集を提供できます。あなたのすべての需要を満たすためには、一緒に努力します。躊躇われずに我々の模試験を利用してみてください。全力を尽くせば、C-S4CPB-2408-JPN試験の合格も可能となります。
The article is outstanding, thank you for sharing! I’m offering the Valid DES-3612 test answers test for free today, as it helped me earn a promotion and raise!
Thank you for your wonderful share; this article really opened my eyes! Free Latest visual PCNSE cert exam shared to elevate your IT skills. Wishing you success in your exams!
I would be very thankful if you continue with quality what you are serving right now with your blog…I really enjoyed it…and i really appreciate to you for this….its always pleasure to read so….Thanks for sharing!!
Actually I read it yesterday but I had some thoughts about it and today I wanted to read it again because it is very well written. s25 ultra manual. I have really enjoyed reading your blog posts.
SAP C_ABAPD_2309인증시험도 어려울 뿐만 아니라 신청 또한 어렵습니다.SAP C_ABAPD_2309시험은 IT업계에서도 권위가 있고 직위가 있으신 분들이 응시할 수 있는 시험이라고 알고 있습니다. 우리 ExamPassdump에서는SAP C_ABAPD_2309관련 학습가이드를 제동합니다. ExamPassdump 는 우리만의IT전문가들이 만들어낸SAP C_ABAPD_2309관련 최신, 최고의 자료와 학습가이드를 준비하고 있습니다. 여러분의 편리하게SAP C_ABAPD_2309응시하는데 많은 도움이 될 것입니다.
Through this article, I gained a lot of useful information. CFM latest soft simulations questions shared for free. Best of luck in your exam preparations!
Can’t resist liking this great content. The CFI-I reliable test lab questions helped me achieve a promotion and salary hike. Now, it’s free for everyone. Best of luck with your professional growth!
The article provided me with a wealth of knowledge. Sharing the AZ-204 valid test sample online test questions with you for free—promotions and salary boosts are within reach!
Thank you for the article; it really piqued my interest. Grab your free Valid exam PSM-I study guide test questions—promotion and salary increases await!
What a remarkable article, I’m so thankful for your share. I’m sharing the New exam CCQM guide files exam materials for free—best of luck!
Die Prüfungsmaterialien von Juniper JN0-637 Zertifizierungsprüfung von unserem It-Pruefung existieren in der Form von PDF und Stimulationssoftware, in der alle Testaufgaben und Antworten von Juniper JN0-637 Zertifizierung enthalten sind. Inhalte dieser Lehrbücher sind umfassend und zuversichtlich. Hoffentlich kann It-Pruefung Ihr bester Hilfer bei der Vorbereitung der Juniper JN0-637 Zertifizierungsprüfung werden. Falls Sie leider die JN0-637 Prüfung nicht bestehen, bitte machen Sie keine Sorge, denn wir werden alle Ihre Einkaufsgebühren bedingungslos zurückgeben.
DumpTOP는 고품질의 IT HP HPE2-B02시험공부자료를 제공하는 차별화 된 사이트입니다. DumpTOP는HP HPE2-B02응시자들이 처음 시도하는HP HPE2-B02시험에서의 합격을 도와드립니다. 가장 적은 시간은 투자하여 어려운HP HPE2-B02시험을 통과하여 자격증을 많이 취득하셔서 IT업계에서 자신만의 가치를 찾으세요.
Oracle的1Z0-1195-25考試認證是業界廣泛認可的IT認證,世界各地的人都喜歡Oracle的1Z0-1195-25考試認證,這項認證可以強化自己的職業生涯,使自己更靠近成功。談到Oracle的1Z0-1195-25考試,KaoGuTi Oracle的1Z0-1195-25的考試培訓資料一直領先於其他的網站,因為KaoGuTi有一支強大的IT精英團隊,他們時刻跟蹤著最新的 Oracle的1Z0-1195-25的考試培訓資料,用他們專業的頭腦來專注於 Oracle的1Z0-1195-25的考試培訓資料。
Fast2test剛剛發布了最新的312-50v13認證考試所有更新的問題及答案,來確保您考試成功通過。我們提供最新的PDF和軟件版本的問題和答案,可以保證考生的312-50v13考試100%通過。在我們的網站上,您將獲得我們提供的ECCouncil 312-50v13免費的PDF版本的DEMO試用,您會發現這絕對是最值得信賴的學習資料。對于擁有高命中率的ECCouncil 312-50v13考古題,還在等什么,趕快下載最新的題庫資料來準備考試吧!
Thank you for your article; it really opened my eyes! Test MB-210 questions answers offers a broad range of material free of charge, hoping it supports you.
it’s really nice and meanful. it’s really cool blog. Linking is very useful thing. You have really helped lots of people who visit blog and provide them usefull information. kihanson.
Your article has made a real difference to me, thank you! These C_C4H63_2411 positive feedback questions were the key to my career boost and salary increase—grab them for free today!
This kind of clever work and reporting! Keep up the terrific works guys I’ve included you guys. download s25 ultra user guide
This kind of clever work and reporting! Keep up the terrific works guys I’ve included you guys. Samsung galaxy a56 review
I would be very thankful if you continue with quality what you are serving right now with your blog…I really enjoyed it. download s25 ultra user guide
No doubts here—liking this content immediately. This is the Reliable 1z0-1054-24 test guide exam that helped me land my promotion and salary raise. It’s free today—wishing you all the best in your career progress!
Your article is exceptional, thank you for sharing! Free access to the 1z0-1127-24 top exam dumps materials, wishing you all the best of luck!
I really appreciate your article, it was so insightful. The 220-1201 reliable test simulator exam is on the horizon. Wish me luck!
さまざまな年齢層の研究条件に基づくさまざまな種類のアンケートによると、当社の300-610テスト準備はこれらの研究グループ向けに完全に設計されており、300-610試験の準備時の能力と効率を向上させ、目標とする300-610証明書が正常に作成されました。 300-610の質問トレントには多くの利点がありますので、ご紹介します。Ciscoの300-610試験に合格することができます。
Your article left me in awe, thank you for sharing! Latest test cram 1Z1-182 sheet offers a wealth of material for free, designed to aid your learning.
This is a truly magnificent article, thank you for sharing your thoughts. Using the AgilePM-Foundation vce files, I achieved a promotion and salary boost. It’s free for everyone now. Wishing you all success in your career paths!
I’m deeply grateful to NaturePath Herbal Clinic for the remarkable improvement in my Amyotrophic Lateral Sclerosis (ALS) condition. Prior to starting their ALS Herbal Treatment program, my health was declining rapidly. After few months on their ALS Herbal treatment program, I’ve seen major progress, I can now walk independently, and my speech has significantly improved. Visit http://www.naturepathherbalclinic.com.
Die Google Associate-Data-Practitioner Zertifizierungsprüfung ist eine Prüfung, die Fachkenntnisse eines Menschen testet. ExamFragen ist eine Website, die Ihnen zum Bestehen der Google Associate-Data-Practitioner Zertifizierungsprüfung verhilft. Vor der Prüfung können Sie die zielgerichteten benutzen, werden Sie in kurz Zeit große Fortschritte machen.
Your blog provided us with valuable information to work with
I was fascinated by the depth of the article. I’m taking the Question APS explanations exam soon. Fingers crossed for success!
I feel so much more enlightened after reading it. Get your hands on the free PC-BA-FBA-20 practice test fee exam questions. Wishing you the best of luck!
Your article is absolutely captivating, thank you for sharing! Using the Valid test dumps 2V0-13.24 questions, I achieved a promotion and salary boost. It’s free for everyone now. Wishing you all success in your career paths!
The content is incredible, I don’t even have to think before liking it. I owe my promotion and raise to the 1Z0-1095-23 accurate study material questions, and today I’m sharing them for free with all of you!
Thank you for your brilliant article; it truly shocked me! Free 156-836 reliable test passing score exam papers available. Best of luck to everyone!
Your article is truly amazing, I appreciate you sharing it. Best of luck! Here are the SailPoint-Certified-IdentityNow-Engineer exam details resources, free for everyone!
Living with Pulmonary Fibrosis (PF) was one of the hardest experiences of my life. The breathlessness, the fatigue, and the fear of the future weighed on me every single day. I had tried so many treatments and medications, but nothing seemed to stop the disease from progressing.Out of both hope and desperation, I came across NaturePath Herbal Clinic. At first, I was skeptical but something about their natural approach and the stories I read gave me the courage to try one more time.I began their herbal treatment program, and within a few weeks, I noticed small changes easier breathing, more energy, and a clearer mind. Over themonths, those improvements became more and more obvious. Today, I can truly say my life has changed. My lungs feel stronger, and my quality of life has returned in ways I didn’t think were possible.This isn’t just a testimony it’s a heartfelt recommendation to anyone struggling with PF or other chronic conditions. Don’t give up hope. I’m so grateful I gave NaturePath Herbal Clinic a chance.
Visit their website to learn more: www. naturepathherbalclinic .com
Living with Pulmonary Fibrosis (PF) was one of the hardest experiences of my life. The breathlessness, the fatigue, and the fear of the future weighed on me every single day. I had tried so many treatments and medications, but nothing seemed to stop the disease from progressing.Out of both hope and desperation, I came across NaturePath Herbal Clinic. At first, I was skeptical but something about their natural approach and the stories I read gave me the courage to try one more time.I began their herbal treatment program, and within a few weeks, I noticed small changes easier breathing, more energy, and a clearer mind. Over themonths, those improvements became more and more obvious. Today, I can truly say my life has changed. My lungs feel stronger, and my quality of life has returned in ways I didn’t think were possible.This isn’t just a testimony it’s a heartfelt recommendation to anyone struggling with PF or other chronic conditions. Don’t give up hope. I’m so grateful I gave NaturePath Herbal Clinic a chance.
Visit their website to learn more: www. naturepathherbalclinic .com
I needs to spend some time learning more or working out more. Thanks for great information I used to be searching for this information for my mission.
Thank you for sharing this truly remarkable article! Upgrade your IT abilities—free Test MB-335 dumps.zip is now available! Best of luck to all!
Your article is a masterpiece, thank you for sharing it! I advanced with the New CRT-211 test simulator online, earning both a promotion and salary increase. Now, it’s free for everyone. Best wishes to all!
I am looking for this informative post thanks for share it
HOW TO RECOVER LOST BTC WITH ZENITH HACKER
We are thrilled to announce that Zenith Hackers Intelligence is now a licensed and certified provider of crypto recovery services! Our specialized team focuses on recovering lost assets from scammers through expert insights in asset recovery and cyber intelligence. If you need assistance with stolen or lost cryptocurrencies, please contact us at +44(755)248-6027 or email zenithintel@consultant.com. We welcome you to visit our office at 100 Preston’s Rd, London E14 9RL, UK for further inquiries.
I sincerely appreciate your share; the content of the article is truly astounding! Get the New IIA-CIA-Part3 test dumps demo test questions for free—your key to the next level in your career!
I was diagnosed with ALS four years ago. For over two years, I relied on prescription medications and therapies, but unfortunately, the symptoms continued to worsen. My mobility declined, muscle weakness increased, and I experienced growing fatigue and discomfort that affected my daily life.
Last year, out of desperation and hope, I decided to try an herbal treatment program from NaturePath Herbal Clinic. Honestly, I was skeptical at first, but within a few months of starting the treatment, I began to notice real changes. My energy improved, the discomfort eased, and I felt stronger and more capable in my daily life.Incredibly, I also regained much of my stamina, balance, and confidence. It has truly been a life-changing experience I feel more like myself again, better than I’ve felt in years. If you or a loved one is struggling with ALS, I sincerely recommend looking into their natural approach. You can visit their website at www. naturepathherbalclinic .com
DumpTOP는 응시자에게 있어서 시간이 정말 소중하다는 것을 잘 알고 있으므로 Cisco 100-140덤프를 자주 업데이트 하고, 오래 되고 더 이상 사용 하지 않는 문제들은 바로 삭제해버리며 새로운 최신 문제들을 추가 합니다. 이는 응시자가 확실하고도 빠르게Cisco 100-140덤프를 마스터하고Cisco 100-140시험을 패스할수 있도록 하는 또 하나의 보장입니다.
This article is exceptional in every way, I’m so thankful for your share. Here’s the free Advanced-Administrator real question on the exam material! Wishing everyone good luck on their exams!
The content of your article is so remarkable, thank you for sharing! Best of luck with your exams—free Pdf C-S4CS-2502 dumps exam papers are available now!
The points mentioned in the article are very useful and have helped me a lot. The 300-815 relevant questions exam is here – wish me luck!
It was a truly rewarding and enlightening piece of writing. Best of luck! Here’s the free Reliable ABMM exam guide files material to help you.
面對職場的競爭和不景氣時期,提升您的專業能力是未來最好的投資,而獲得OCEG GRCP認證對于考生而言有諸多好處。相對于考生尋找工作而言,一張GRCP認證可以倍受企業青睞,為您帶來更好的工作機會。但是如何輕松拿到GRCP認證哪? NewDumps的GRCP考古題是通過考試最有效的方式之一,我們提供在線測試引擎的題庫,可以讓您模擬真實的考試情景,快速讓考生掌握知識點并應用。GRCP題庫資料包含真實的考題體型,100%幫助考生通過考試。
Fast2test的專家團隊利用他們的經驗和知識終於研究出了關於ISACA CISM-CN 認證考試的培訓資料。我們的ISACA CISM-CN 認證考試培訓資料很受客戶歡迎,這是Fast2test的專家團隊勤勞勞動的結果。他們研究出來的模擬測試題及答案有很高的品質,和真實的考試題目有95%的相似性,是很值得你依賴的。如果你使用了Fast2test的培訓工具,你可以100%通過你的第一次參加的ISACA CISM-CN認證考試。
What an insightful article, I truly appreciate your sharing it! Free Latest study guide 300-715 ebook test papers for everyone—promotions and salary raises await!
What an incredible article, thank you for sharing this inspiration! The free Latest CPMAI_v7 associate level exam test materials are available for all. Good luck!
This article is absolutely amazing, thank you for sharing! Get access to the DVA-C02 valid exam fee test that was essential to my promotion and salary raise. It’s free for you today—best of luck!
I was amazed by the insights in that article. The C-BCHCM-2502 latest dumps free download exam is almost here. Hope I ace it!
I’m inspired by the message in this article, thank you for sharing! Bang King 15000 Disposable Vape | 15K Puffs, Multiple Nicotine Strengths – Wholesale and Bulk Buy for Disposable Vapes
What a captivating article, I truly appreciate your sharing it. New practice questions C1000-182 files provides great resources for free, and I hope you find them useful.
I’m absolutely sold on this content, liking it now. The Latest CCSFP test prep exam questions are free—take the first step towards career advancement and salary growth!
Pass4Test의 SAP C-THR84-2505덤프는 SAP C-THR84-2505시험문제변경에 따라 주기적으로 업데이트를 진행하여 덤프가 항상 가장 최신버전이도록 업데이트를 진행하고 있습니다.구매한 SAP C-THR84-2505덤프가 업데이트되면 저희측에서 자동으로 구매시 사용한 메일주소에 업데이트된 최신버전을 발송해드리는데 해당 덤프의 구매시간이 1년미만인 분들은 업데이트서비스를 받을수 있습니다.
Thank you for the article, it really brought a fresh perspective. Wga Crystal Plus 20000 Puffs: Engangs-vape med dobbelt smag og skærm – Engros- og massekøb af engangs-vape
This article is a real treasure, thank you for sharing it with us. Here are the New CIC test lab questions materials, free of charge—best of luck!
This is truly a magnificent article, thank you for sharing. AlFakher Crown Bar Pro 15000 Puffs одноразовий вейп з зарядкою USB-C – Купити одноразові вейпи оптом і в роздріб
I’m truly thankful for your article, it left a lasting and meaningful impression. Elfbox RGB 14000 Pro engångsvape – 14000 puffar, nätspole – grossist- och bulkköp för engångsvape
Your article was truly refreshing, thank you so much. The API-571 reliable exam passing score exam is approaching. Hope I ace it!
Truly an exceptional piece of writing, thank you for sharing. RandM Tornado 15000 Engangs-vape – 15K pust, genopladelig, 25ml – Engros- og massekøb af engangs-vape
KoreaDumps는 한국어로 온라인상담과 메일상담을 받습니다. Fortinet FCSS_SOC_AN-7.4덤프구매후 일년동안 무료업데이트서비스를 제공해드리며Fortinet FCSS_SOC_AN-7.4시험에서 떨어지는 경우Fortinet FCSS_SOC_AN-7.4덤프비용 전액을 환불해드려 고객님의 부담을 덜어드립니다. 더는 고민고민 하지마시고 덤프 받아가세요.
The content is awesome; I’ll like it right now. “IREX TWINS 60000 Disposable Vape” – 40 000 pufų, dvigubas bakas, 5% nikotino – didmeninė ir didmeninė prekyba vienkartiniais garintuvais
Thank you for your article, it was a true eye-opener. Enjoy these free PostgreSQL-Essentials valid exam dumps free materials. Good luck with your exam!
What a remarkable article, I truly appreciate you sharing it. Free FAAA_005 vce download exam resources are available now—good luck!
您選擇我們的Fast2test來幫助你通過Salesforce Plat-101 認證考試試是一個明智的選擇。你可以先線上免費下載Fast2test為你提供的關於Salesforce Plat-101 認證考試練習題及答案的試用版本作為嘗試,那樣你會更有信心選擇我們Fast2test的產品來準備Salesforce Plat-101 認證考試。如果你考試失敗,我們會全額退款給你。
This is an exceptional article, thank you for sharing this insight. OKSO Shisha Hookah Mega 50000 使い捨てベイプ – 40ml Eリキッド、50Kパフ – 使い捨てベイプの卸売り・まとめ買い
This article is truly awe-inspiring, thank you for sharing! OKSO Double Flavor 60000 Puffs Disposable Vape – Dual Taste, Mesh Coil – Wholesale and Bulk Buy for Disposable Vapes
Thank you for bringing such an extraordinary article! Get access to the Latest COF-C02 test cram test that was essential to my promotion and salary raise. It’s free for you today—best of luck!
Liking this instantly, it’s that good! Zooy Twins 35000 Dual Flavor Engangs-vape – 35.000 pust – Engros- og massekøb af engangs-vape
Truly a brilliant article, I’m so appreciative of your sharing. This New 156-587 test questions fee set helped me earn a promotion and raise. Now, I’m offering it for free. Best of luck with your career success!
This article is truly top-notch, thank you for sharing it with me. Here’s the ISO-45001-Lead-Auditor valid study guide files exam that boosted my career with a promotion and salary increase. It’s free today for all! Wishing you success in your career journey.
Hi, thanks for sharing this blog with us very nice and informative post . Regards
Visa Consultancy
I sincerely thank you for sharing; the article is truly fantastic! The 1Z0-1145-1 valid test collection pdf exam that helped me get promoted and earn a salary raise is available for you today at no charge. Hope you reach your career targets soon!
Your article is truly stunning; I appreciate your sharing! Best of luck! Sharing the Valid study guide UiPath-SAIAv1 sheet materials with you all.
This research on restoring SMN expression in SMA through targeting SR proteins is fascinating! For anyone interested in exploring more about innovative therapies and molecular mechanisms, you might want to check out opportunities to write for us technology. It’s a great way to share insights on cutting-edge biomedical advances.
Truly a remarkable read, thank you for sharing this brilliant article. I got promoted and earned a raise using this JN0-750 valid exam questions answers. Now it’s available for free. Best of luck with your promotions!
Thank you for sharing; it was truly incredible! The Reliable study questions 156-587 ebook content is excellent, and you can access it for free.
Truly an impressive article, I’m grateful for your share. Sharing the Reliable ACD201 study notes test that contributed to my promotion and salary hike, and it’s free for you to access today. Best of luck in achieving your career aspirations!
I’m truly grateful for your article, it left an indelible mark. The Latest IIA-CIA-Part3 braindumps files resource is packed with content and made available for free to help you.
Your article was amazing, I’m truly grateful for it. Upgrade your IT knowledge and access 300-415 latest test lab questions for free. Wishing you the best of luck!
You write well I really like what you write.
I always look forward to your posts! Really great read!
Many viewpoints in the article have been very inspiring, and I’ve benefited greatly. We’re providing Exam D-DS-FN-23 sims, filled with valuable content, for free to support you.
Die Sicherheitsprotokolle von zoome casino test , insbesondere die Zwei-Faktor-Authentifizierung für Konten, sind ein Bereich, den ich als besonders wichtig für ein deutsches Echtgeld-Casino 2025 erachte. Die Verschlüsselung der Datenübertragung scheint dem neuesten Stand der Technik zu entsprechen, und die Tatsache, dass sie offensiv über ihre Sicherheitsmaßnahmen informieren, schafft eine psychologische Sicherheit, die für das Einzahlen großer Summen notwendig ist.
Your article was so impressive, thank you for sharing! Official GES-C01 practice test helped me get ahead in my career and earn more. Now, I’m offering it to you for free!
hi, thanks for sharing this with us . very nice and informative blog . Best regards
visa consultants
This article provides a thoughtful and informative overview of the escort scene in Ajman, highlighting professionalism, discretion, and respectful interactions. I appreciate the focus on selecting reputable and licensed services, as well as emphasizing safety and clear communication—elements often overlooked in discussions about this topic. Content like this helps readers gain a responsible and practical understanding of the industry while avoiding sensationalism. By stressing ethical practices and mutual respect, it adds meaningful value for anyone looking to approach this subject responsibly. Thank you for sharing this insightful post—it encourages awareness, responsibility, and respectful engagement.
This article provides a well-rounded and informative perspective on the call girl industry in Dubai, highlighting professionalism, discretion, and respectful interactions. I appreciate the emphasis on choosing reputable and licensed services, as well as prioritizing safety and clear communication—points often overlooked in discussions about this topic. Content like this helps readers develop a responsible and practical understanding of the industry while avoiding sensationalism. By stressing ethical practices and mutual respect, it adds meaningful value for anyone seeking insight into this subject. Thank you for sharing this thoughtful post—it encourages awareness, responsibility, and respectful engagement.
This article provides a well-rounded and informative overview of the escort industry in Abu Dhabi, highlighting professionalism, discretion, and respectful interactions. I appreciate the focus on choosing reputable and licensed services, as well as prioritizing safety and clear communication—points often overlooked in discussions about this topic. Content like this helps readers gain a responsible and practical understanding of the industry while avoiding sensationalism. By emphasizing ethical behavior and mutual respect, it adds meaningful value for anyone looking to approach this subject responsibly. Thank you for sharing this thoughtful post—it encourages awareness, responsibility, and respectful engagement.
This article provides a thoughtful and informative perspective on the escort scene in Ajman, highlighting professionalism, discretion, and respectful interactions. I appreciate the focus on selecting reputable and licensed services, as well as prioritizing safety and clear communication—points often overlooked in discussions on this topic. Content like this helps readers develop a responsible and practical understanding of the industry while avoiding sensationalism. By emphasizing ethical practices and mutual respect, it adds real value for anyone looking to approach this subject responsibly. Thank you for sharing this insightful post—it encourages awareness, responsibility, and respectful engagement.
This article provides a well-rounded and informative perspective on the escort industry in Sharjah, highlighting professionalism, discretion, and respectful interactions. I appreciate the focus on selecting reputable and licensed services, as well as prioritizing safety and clear communication—points often overlooked in discussions about this topic. Content like this helps readers gain a responsible and practical understanding of the industry while avoiding sensationalism. By stressing ethical practices and mutual respect, it adds meaningful value for anyone looking to approach this subject responsibly. Thank you for sharing this thoughtful post—it encourages awareness, responsibility, and respectful engagement.
This article provides a balanced and informative perspective on Indian companions in JBR, highlighting professionalism, discretion, and respectful interactions. I appreciate the emphasis on choosing reputable and licensed services, as well as prioritizing safety and clear communication—points often overlooked in discussions on this topic. Content like this helps readers develop a responsible and practical understanding of the industry while avoiding sensationalism. By stressing ethical practices and mutual respect, it adds real value for anyone seeking insight into this subject responsibly. Thank you for sharing this thoughtful post—it encourages awareness, responsibility, and respectful engagement.
This article provides a well-rounded and informative perspective on Dubai escort services, highlighting professionalism, discretion, and respectful interactions. I appreciate the emphasis on choosing reputable and licensed services, as well as prioritizing safety and clear communication—elements often overlooked in discussions about this topic. Content like this helps readers develop a responsible and practical understanding of the industry while avoiding sensationalism. By stressing ethical practices and mutual respect, it adds meaningful value for anyone looking to approach this subject responsibly. Thank you for sharing this insightful post—it encourages awareness, responsibility, and thoughtful engagement.
This article provides a well-informed and balanced perspective on the escort industry in Dubai, highlighting professionalism, discretion, and respectful interactions. I appreciate the focus on selecting reputable and licensed services, as well as prioritizing safety and clear communication—elements often overlooked in discussions about this topic. Content like this helps readers develop a responsible and practical understanding of the industry while avoiding sensationalism. By emphasizing ethical practices and mutual respect, it adds meaningful value for anyone seeking insight into this subject. Thank you for sharing this thoughtful post—it encourages awareness, responsibility, and respectful engagement.
This article provides a well-rounded and informative perspective on call girls in Abu Dhabi, highlighting professionalism, discretion, and respectful interactions. I appreciate the emphasis on choosing reputable and licensed services, as well as prioritizing safety and clear communication—points often overlooked in discussions about this topic. Content like this helps readers develop a responsible and practical understanding of the industry while avoiding sensationalism. By stressing ethical practices and mutual respect, it adds meaningful value for anyone looking to approach this subject responsibly. Thank you for sharing this thoughtful post—it encourages awareness, responsibility, and respectful engagement.
This article provides a clear and informative perspective on call girls in Ajman, highlighting professionalism, discretion, and respectful interactions. I appreciate the focus on selecting reputable and licensed services, as well as prioritizing safety and clear communication—elements often overlooked in discussions about this topic. Content like this helps readers develop a responsible and practical understanding of the industry while avoiding sensationalism. By emphasizing ethical behavior and mutual respect, it adds real value for anyone looking to approach this subject responsibly. Thank you for sharing this insightful post—it encourages awareness, responsibility, and thoughtful engagement.
This article provides a balanced and informative perspective on call girls in Sharjah, highlighting professionalism, discretion, and respectful interactions. I appreciate the emphasis on choosing reputable and licensed services, as well as prioritizing safety and clear communication—elements often overlooked in discussions about this topic. Content like this helps readers develop a responsible and practical understanding of the industry while avoiding sensationalism. By stressing ethical practices and mutual respect, it adds meaningful value for anyone seeking insight into this subject. Thank you for sharing this thoughtful post—it encourages awareness, responsibility, and respectful engagement.
This article provides a well-rounded and informative perspective on Indian escorts in Dubai, highlighting professionalism, discretion, and respectful interactions. I appreciate the emphasis on choosing reputable and licensed services, as well as prioritizing safety and clear communication—points often overlooked in discussions on this topic. Content like this helps readers develop a responsible and practical understanding of the industry while avoiding sensationalism. By stressing ethical practices and mutual respect, it adds meaningful value for anyone looking to approach this subject responsibly. Thank you for sharing this insightful post—it encourages awareness, responsibility, and thoughtful engagement.
Nun ist die PMI CPMAI Zertifizierungsprüfung eine beliebte Prüfung in der IT-Branche. Viele IT-Fachleute wollen das PMI CPMAI Zertfikat erhalten. So ist die PMI CPMAI Zertifizierungsprüfung eine beliebte Prüfung. Das PMI CPMAI Zertfikat ist sehr hilfreich, um Ihre Arbeit in der IT-Industrie zu verbessern und Ihr Gehalt zu erhöhen und Ihrem Leben eine zuverlässige Garantie zu geben.
Your article was so impressive, I’m deeply thankful. Latest exam camp Salesforce-MuleSoft-Associate free test questions—free for all. Wishing you the best of luck in your exams!
NewDumps為Microsoft SC-200 認證考試準備的培訓包括Microsoft SC-200認證考試的模擬測試題和當前的考試真題。在互聯網上你也可以看到幾個也提供相關的培訓的網站,但是你比較之後,你就會發現NewDumps的關於Microsoft SC-200 認證考試的培訓比較有針對性,不僅品質是最高的,而且內容是最全面的。
I’ve been really interested in reading the posts on this regularly updated website. There’s a lot of great content here. Just wanted to say that I really enjoyed this post. It’s really great. Please keep it up.
The progression of ideas in this article is excellent. Each section connects well with the next, making it easy to stay engaged and retain the information
Thank you so much for such a well-written article. It’s full of insightful information. Your point of view is the best among many without fail. For certain, It is one of the best blogs in my opinion.
I really appreciate it. with this great post you gave us. I will visit often.
I love how you package different topics into something interesting and informative. Do you have any advice for someone just starting a blog?
I really appreciate your courage in presenting this new perspective. It opened my eyes to things I had never thought about before
I needs to spend some time learning more or working out more. Thanks for great information I used to be searching for this information for my mission.
Wonderful article Thanks for sharing the information with us,
Ready to level up your Azure skills? The AZ-204 certification opens doors to advanced cloud development roles. With expert prep resources, real exam questions, and practical tips, you’ll boost your chances of passing on the first try. Start your journey today and unlock new career opportunities!
https://www.marks4sure.co/AZ-204-exam.html
The text addresses the topic in a simple way. Ideas are understandable but lack development. The learner needs to improve grammar, spelling, and sentence structure. Practice and revision are necessary to enhance writing quality.
This text shows a good attempt to express ideas clearly. The learner uses simple but effective language. Grammar and spelling need some revision, yet the overall meaning is clear. Continued effort will lead to better results.
The learner demonstrates a satisfactory level of writing. The ideas are clear and related to the topic. The text is organized in a simple but logical way. Some grammatical and spelling mistakes are present, yet the overall meaning remains clear. More practice will help improve accuracy.
The learner presents relevant ideas related to the topic. The text is generally clear and organized. Vocabulary is suitable and sentences are mostly correct. Some mistakes remain, but they do not prevent understanding. More practice will help improve accuracy.
Thank you for sharing this valuable update. The article is informative and easy to follow. I appreciate the effort behind producing quality content.
Thank you for consistently offering content that supports learning and growth. This article explains concepts clearly and thoroughly. I appreciate the ongoing effort behind your updates.
I’ve read some good stuff here. Definitely worth bookmarking for revisiting. I surprise how much effort you put to create such a great informative website. Schwimmbadbauer
We are tied directly into the sate’s renewal database which allows us to process your request almost instantly. Batterij 10 kWh
I was very pleased to find this site.I wanted to thank you for this great read!! I definitely enjoying every little bit of it and I have you bookmarked to check out new stuff you post. Polypropylen Schwimmbecken
This is truly an practical and pleasant information for all. Thanks for sharing this to us and more power Aanleg buitenzwembad
I have been searching to find a comfort or effective procedure to complete this process and I think this is the most suitable way to do it effectively. Gartenpools
The worst part of it was that the software only worked intermittently and the data was not accurate. You obviously canot confront anyone about what you have discovered if the information is not right. Batterij 15 kWh
It proved to be Very helpful to me and I am sure to all the commentators here! Schwimmbecken Garten
The next time I read a blog, I hope that it doesnt disappoint me as much as this one. I mean, I know it was my choice to read, but I actually thought you have something interesting to say. All I hear is a bunch of whining about something that you could fix if you werent too busy looking for attention. Zwembad plaatsen tuin
I just found this blog and have high hopes for it to continue. Keep up the great work, its hard to find good ones. I have added to my favorites. Thank You. Polypropylen pools
Thanks for sharing this information. I really like your blog post very much. You have really shared a informative and interesting blog post with people.. BYD thuisbatterij
Took me time to understand all of the comments, but I seriously enjoyed the write-up. It proved being really helpful to me and Im positive to all of the commenters right here! Its constantly nice when you can not only be informed, but also entertained! I am certain you had enjoyable writing this write-up. Kosten Pool
I truly like you’re composing style, incredible data, thankyou for posting. Prijs zwembad
It’s really nice and meanful. it’s really cool blog. Linking is very useful thing.you have really helped lots of people who visit blog and provide them usefull information. LPW Pools
I definitely enjoying every little bit of it and I have you bookmarked to check out new stuff you post. Aanleg zwembad tuin
Nice blog and absolutely outstanding. You can do something much better but i still say this perfect.Keep trying for the best. Swimmingpools
Your article is a work of art, thank you for sharing it with us! I’m sharing the Certification Marketing-Cloud-Email-Specialist dump questions that helped me get promoted and earn more—grab them for free!
wild that smn2 can almost do the job but misses because of one single nucleotide in exon 7
I liked your article and I hope you will have many entries or more Fox ESS thuisbatterij
Very interesting blog. Alot of blogs I see these days don’t really provide anything that I’m interested in, but I’m most definately interested in this one. Just thought that I would post and let you know. Goodwe thuisbatterij prijs
I was surfing net and fortunately came across this site and found very interesting stuff here. Its really fun to read. I enjoyed a lot. Thanks for sharing this wonderful information. JA Solar zonnepanelen
That is very helpful for increasing my knowledge in this field. Goodwe thuisbatterij
Great post I would like to thank you for the efforts you have made in writing this interesting and knowledgeable article. Huawei thuisbatterij 5 kwh
Wow, happy to see this awesome post. I hope this think help any newbie for their awesome work. By the way thanks for share this awesomeness from Airco
Took me time to understand all of the comments, but I seriously enjoyed the write-up. It proved being really helpful to me and Im positive to all of the commenters right here! Its constantly nice when you can not only be informed, but also entertained! I am certain you had enjoyable writing this write-up. BYD thuisbatterij
I read your post and I found it amazing! thank! Jinko zonnepanelen
Nice blog. Found this while searching through Huawei thuisbatterij
I’ve read some good stuff here. Definitely worth bookmarking for revisiting. I surprise how much effort you put to create such a great informative website. Zonnepanelen Qcells
It’s late finding this act. At least, it’s a thing to be familiar with that there are such events exist. I agree with your Blog and I will be back to inspect it more in the future so please keep up your act. Sma thuisbatterij
If you don”t mind proceed with this extraordinary work and I anticipate a greater amount of your magnificent blog entries REC zonnepanelen
I am very enjoyed for this blog. Its an informative topic. It help me very much to solve some problems. Its opportunity are so fantastic and working style so speedy. SolarEdge thuisbatterij
I really enjoyed reading this post, big fan. Keep up the good work andplease tell me when can you publish more articles or where can I read more on the subject? Sunpower Maxeon zonnepanelen
I feel very grateful that I read this. It is very helpful and very informative and I really learned a lot from it. Enphase batterij
wild how one single nucleotide in exon 7 makes such a massive difference between SMN1 and SMN2.
So SMN2 is basically just a broken backup because of that single nucleotide in exon7.
Thank you for sharing this amazing article, it is truly breathtaking! Best of luck to everyone! Free Valid Terraform-Associate-003 vce test simulator questions are now available.
This article is amazing, thank you for sharing it with us! Sharing free Detailed Cybersecurity-Practitioner answers study materials. Wishing you the best of luck!
Thank you for sharing such informative content. The article is easy to understand and offers helpful knowledge. I appreciate the effort behind maintaining this educational website.
I appreciate the helpful knowledge provided in this post. The explanations are simple and easy to understand. Thank you for offering content that truly benefits readers.
I’m very grateful for your article, it really caught my attention. The AP-226 latest exam objectives pdf resources are highly valuable and free.
Als ich begonnen habe, mich intensiver mit digitalen Unterhaltungsplattformen zu beschäftigen, war mir schnell klar, dass ich mehr suche als nur ein großes Angebot an Spielen. Für mich zählt vor allem, wie angenehm sich die gesamte Nutzung anfühlt – vom ersten Öffnen der Seite bis zur Auswahl meiner Favoriten. Genau deshalb habe ich mich näher mit trip2vip casino beschäftigt und festgestellt, dass eine moderne Plattform nicht nur technisch stabil laufen muss, sondern auch ein durchdachtes Konzept braucht. Ich mag es, wenn Inhalte logisch strukturiert sind und ich innerhalb weniger Sekunden zwischen verschiedenen Kategorien wechseln kann, ohne Zeit mit unnötigem Suchen zu verlieren. Besonders wichtig ist mir außerdem, dass neue Inhalte regelmäßig ergänzt werden, denn ich probiere gern unterschiedliche Spielmechaniken aus und entdecke gerne innovative Funktionen. Wenn dann noch schnelle Ladezeiten und ein unkompliziertes Zahlungsmanagement dazukommen, entsteht für mich genau die Art von digitalem Erlebnis, die ich nach einem langen Arbeitstag suche – entspannt, abwechslungsreich und komfortabel, ohne überladen oder kompliziert zu wirken.
The content is excellent, so I’m liking it now. With the help of this H19-308_V4.0 trustworthy dumps, I earned a promotion and a pay raise. Now I’m offering it for free to everyone. Wishing you all success in your careers!
Wild that a single nucleotide in exon 7 is basically the difference between a functioning system and Type I SMA.
It’s wild how much that one nucleotide in exon 7 messes with the SMN2 splicing.
Perfectly executed content, like button pressed. I owe my success to the MS-102 new test bootcamp materials exam, and now I’m giving you the chance to access it for free!
Thank you for your article, it was a real revelation to me. It’s time for the Archer-Expert best vce exam! Wish me good fortune!
I really like reading through a post that can make men and women think. Also, thank you for allowing me to comment
Hi there to all, for the reason that I am genuinely keen of reading this website’s post to be updated on a regular basis. It carries pleasant stuff.
Amazing stuff here, definitely hitting that like. Unlock the door to promotions and salary raises with the free Vce Arch-302 files questions!
Offermaidsprovides reliable and professional cleaning services in Dubai with trained staff and affordable pricing. Perfect choice for busy families looking for quality home cleaning and maid services. Highly recommended!
Thank you for this amazing article, it left a profound impression on me. Here’s the High C1000-166 passing score test that helped me land my promotion and raise. It’s free today—good luck with your career progress!
The depth and scope of the article opened my eyes. XSOAR-Engineer reliable test questions fee offers a wealth of material for free, designed to aid your learning.
I am genuinely impressed by your unique perspective and the depth of insight you bring, as your writing not only informs but also inspires readers to think more deeply, leaving a memorable and positive impression long after finishing the article.
I sincerely admire the elegance of your writing style, as each paragraph is thoughtfully developed and rich with insight, creating an article that feels informative, engaging, and rewarding for readers seeking quality content.
Your ability to communicate complex ideas is impressive because you simplify difficult concepts effectively while still maintaining depth and accuracy in your writing which shows your strong understanding of the topic.
I truly respect how your article presents meaningful ideas with such clarity and refinement, making even detailed concepts easy to understand while maintaining a tone that feels both professional and inviting throughout.
I always look forward to your posts! Really great read!
Great Idea. I appreciate the insights you’ve shared. heng36
I liked your article and I hope you will have many entries or more Airco installatie Leuven
I have bookmarked your website because this site contains valuable information in it. I am really happy with articles quality and presentation. Thanks a lot for keeping great stuff. I am very much thankful for this site. Prijs zonnepanelen
Thank you very much for sharing such a useful article. Will definitely saved and revisit your site Zonnepaneeel installateur
Three are usually cheap Ralph Lauren available for sale each and every time you wish to buy. Zonnepanelen met batterij
Great post I would like to thank you for the efforts you have made in writing this interesting and knowledgeable article. Zonnepaneel installateur limburg
There is definately a great deal to know about this subject. I like all of the points you’ve made. Zonnepaneel installateur limburg
Thanks for sharing this information. I really like your blog post very much. You have really shared a informative and interesting blog post . Warmtepompen Leuven
Hi to everybody, here everyone is sharing such knowledge, so it’s fastidious to see this site, and I used to visit this blog daily Zonnepaneel installateur limburg
I have to search sites with relevant information on given topic and provide them to teacher our opinion and the article. Airconditioning
Thanks for sharing this information. I really like your blog post very much. You have really shared a informative and interesting blog post . Zonnepanelen
This post is good enough to make somebody understand this amazing thing, and I’m sure everyone will appreciate this interesting things. Zonnepaneel installateur
Please give some advice on how to achieve this kind of posts. Warmtepomp
Three are usually cheap Ralph Lauren available for sale each and every time you wish to buy. Lucht-lucht warmtepomp
Thanks for sharing this information. I really like your blog post very much. You have really shared a informative and interesting blog post . Zonnepanelen met batterijen
Hey, great blog, but I don’t understand how to add your site in my rss reader. Can you Help me please? Warmtepompen lucht-lucht
我々社のPalo Alto Networks NetSec-Analyst問題集を購入するかどうかと疑問があると、弊社JPNTestのNetSec-Analyst問題集のサンプルをしてみるのもいいことです。試用した後、我々のNetSec-Analyst問題集はあなたを試験に順調に合格させると信じられます。なぜと言うのは、我々社の専門家は改革に応じて問題の更新と改善を続けていくのは出発点から勝つからです。
I’m so grateful for the detailed explanations in your articles—they’ve helped me learn so much.
Thank you for making complex ideas so easy to understand through your amazing content.
Thank you for sharing excellent articles through your professional and inspiring website. The content is always useful, engaging, and easy to follow. Your hard work and passion for education help readers stay informed and motivated while discovering valuable insights and meaningful knowledge through your online platform daily.
Your transitions between paragraphs are smooth and effective, creating a cohesive structure that guides readers naturally through the discussion without confusion. This article is a wonderful example of writing that successfully combines depth, elegance, and readability in a truly impressive way.
I truly respect how your article presents meaningful ideas with such clarity and refinement, making even detailed concepts easy to understand while maintaining a tone that feels both professional and inviting throughout.
making even detailed concepts easy to under
This blog is so nice to me. I will keep on coming here again and again. LockDown168
You are sharing a very informative and great post.
I appreciate the thoughtful information shared in this article. It is clear, practical, and beneficial for readers seeking reliable knowledge. Thank you for your ongoing dedication to providing helpful updates.
I’m thankful for the valuable knowledge explained here. The writing is concise yet informative, making it easy to understand important points. Thank you for producing such helpful articles.
Choosing verified free credit offers helps players avoid unreliable websites and focus on legitimate promotions.
I adore your websites way of raising the awareness on your readers. Zwembad plaatsen
Interesting post. I Have Been wondering about this issue, so thanks for posting. Pretty cool post.It ‘s really very nice and Useful post.Thanks Polypropylen pools
Great post! I am actually getting ready to across this information, is very helpful my friend. Also great blog here with all of the valuable information you have. Keep up the good work you are doing here. Monoblock zwembad
I have a mission that I’m just now working on, and I have been at the look out for such information Prijs monoblock zwembad
A debt of gratitude is in order for sharing the information, keep doing awesome… I truly delighted in investigating your site. great asset… Polypropyleen zwembad
I finally found great post here.I will get back here. I just added your blog to my bookmark sites. thanks.Quality posts is the crucial to invite the visitors to visit the web page, that’s what this web page is providing. Polypropyleen zwembad
Great Information sharing .. I am very happy to read this article .. thanks for giving us go through info.Fantastic nice. I appreciate this post. Zwembadbouw
Your site is truly cool and this is an extraordinary moving article. Zwembadbouw
Interesting post. I Have Been wondering about this issue, so thanks for posting. Pretty cool post.It ‘s really very nice and Useful post.Thanks Bouwkundig zwembad vs monoblock zwembad
I think this is an informative post and it is very useful and knowledgeable. therefore, I would like to thank you for the efforts you have made in writing this article. Prijs zwembad
I would like to thank you for the efforts you have made in writing this article. I am hoping the same best work from you in the future as well. In fact your creative writing abilities has inspired me to start my own Blog Engine blog now. Really the blogging is spreading its wings rapidly. Your write up is a fine example of it. Polypropyleen zwembad
I really loved reading your blog. It was very well authored and easy to understand. Unlike other blogs I have read which are really not that good.Thanks alot! Inbouwzwembad
Mmm.. good to be here in your article or post, whatever, I think I should also work hard for my own website like I see some good and updated working in your site. Inbouwzwembad
I feel very grateful that I read this. It is very helpful and very informative and I really learned a lot from it. Zwembad tuin kostprijs
I think this is an informative post and it is very useful and knowledgeable. therefore, I would like to thank you for the efforts you have made in writing this article. Zwembad aanleggen in de tuin
https://www.online5numbersummarycalculator.com
This website provides a statistics tool for calculating the five-number summary quickly and clearly. It is useful for students, educators, and anyone working with data analysis or classroom statistics. The tool helps simplify descriptive statistics in an easy online format.
Great overview of the SMA mechanism and the therapeutic logic behind boosting SMN expression through SR proteins. The way you connect exon 7 splicing, SMN1/SMN2 differences, and motor neuron loss makes the article very approachable without losing scientific depth. I especially appreciated the focus on how targeted splicing modulation could translate into real clinical benefit for Type I patients. For readers who like visualizing complex biology, the Image to Line Art Converter is also a nice quick tool to explore.
I am happy to find this post very useful for me, as it contains lot of information. I always prefer to read the quality content and this thing I found in you post. Thanks for sharing