Heather Geist and Juliana Schneider
Summary of:
Wee, CD, Havens, MA, Jodelka, FM, Hastings, ML. (2014). Targeting SR Proteins Improves SMN Expression in Spinal Muscular Atrophy Cells. PLoS ONE, 9(12). e115205. doi:10.1371/journal.pone.0115205
Background:
What is SMA?
Spinal muscular atrophy (SMA) is an autosomal recessive genetic disease that is one of the most common inherited causes of pediatric mortality. SMA has occurs in 1 in 6000 live births and has a carrier frequency of 1 in 40. This disease affects the voluntary muscle movement controls of the nervous system. SMA is categorized as a degenerative neuromuscular disease, which involves the loss of motor neurons in the spinal cord. This in turn prevents the muscle from receiving signals, leading to muscular atrophy over time. The most common form of SMA is called chromosome 5 SMA, which has great variability in age of onset. For this reason it is typically classified into types 1 through 4. Our main focus will be on Type I SMA patients. Type I is correlated with the lowest level of functioning patients. Onset of symptoms in Type I patients usually occurs at birth or during infancy, leading to the greatest impact on motor function. The most drastically affected muscles are typically those closest to the center of the body and because of early onset, these individuals have high mortality rates.
What is SMA Caused by?
SMA is caused by deletions or mutations in the Survival of Motor Neuron I gene (SMN1) located on chromosome 5. SMN proteins are essential for conventional motor neuron function. If a deletion or mutation of SMN1 occurs, an unstable form of the SMN protein results. The SMN2 gene, also located on chromosome 5, has the ability to produce SMN protein at smaller volumes (Figure 1). The main aberration between SMN1 and 2 is the single nucleotide in exon7, which is thought to be an exon splice enhancer. As a result of this single nucleotide difference, SMN2 cannot compensate for the loss of SMN1. The SMN1 gene is telomeric. It contains more than 4 genes, so it is susceptible to rearrangements and deletions. SMN2, however, is centromeric and is not as prone to rearrangement. Interestingly, increasing the inclusion of exon7 escalates the abundance of SMN protein. The SMN2 gene, also located on chromosome 5, produces SMN protein at smaller volumes. Inclusion of exon7 has been shown to have efficacy in animal models of SMA and early human clinical trials.
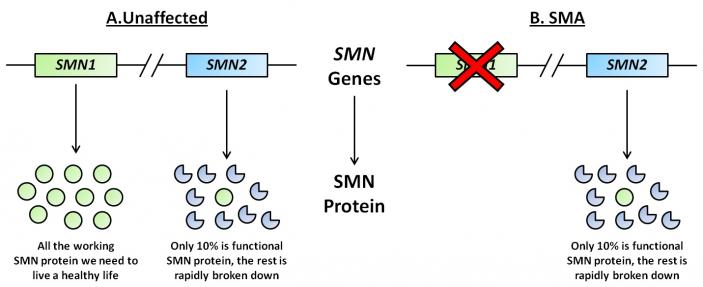 Figure 1. Representative diagram of SMN1 and SMN2 genes and subsequent proteins in unaffected and SMA individuals. (Image modified from http://www.smasupportuk.org.uk/blog/ research/azzouz-laboratory-gene-therapy-for-sma).
Figure 1. Representative diagram of SMN1 and SMN2 genes and subsequent proteins in unaffected and SMA individuals. (Image modified from http://www.smasupportuk.org.uk/blog/ research/azzouz-laboratory-gene-therapy-for-sma).
Splicing and its role in SMA:
Exons and introns undergo a period of pre-mRNA splicing. This is conducted by the spliceosome complex, which is comprised of 5 snRNPs and other splicing proteins/factors (Figure 2). The splicesome complex works to identify exons and introns through binding to specific consensus splicing sequences on both the 5’ and 3’ ends of mRNA. Multiple proteins and sequence elements have been shown to play a role in regulating the splicing of SMN exon7. Specifically, SRSF1, SRSF2, and SRSF9 are known to influence exon7 inclusion, but few other members of the SR protein family have been explored regarding their role in SMN2 exon7 splicing. In addition to the SR protein family, the hnRNP proteins have also been implicated in play a role in exon 7 inclusion or exclusion.
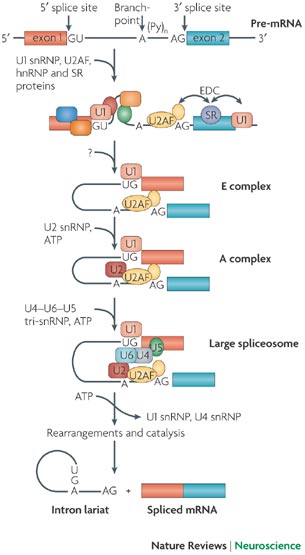 Figure 2. The spliceosome complex and process of pre-mRNA processing. (Image modified from http://www.nature.com/nrn/journal/v8/n11/box/nrn2237_BX1.html)
Figure 2. The spliceosome complex and process of pre-mRNA processing. (Image modified from http://www.nature.com/nrn/journal/v8/n11/box/nrn2237_BX1.html)
Question:
What characteristics and roles do SR and hnRNP proteins play in SMA disease pathogenesis? Do any of these proteins have therapeutic implications?
Experimental Approach:
Two main cell-lines were utilized throughout the experiments. The first was HeLa cells, which are derived from a cervical cancer tumor. The cells were treated and transfected with siRNA and Lipofectamine to knockdown specific SR and hnRNPs. In order to overexpress these proteins, expression vectors were transfected into the cells. The second cell-line used were human fibroblast cells derived from a Type I SMA patient only having one copy of SMN2. These cells were transfected as the HeLa cells were to knockdown the designated SR and hnRNP proteins. After cells were treated, RNA was isolated, and RT-PCR was conducted.
Proteins were extracted from the cells and immunoblots were performed by probing with 12 different antibodies directed at specific proteins. After quantitative analysis of fluorescence was performed, different levels of exon7 expression were observed. In vitro transcription was conducted on RNA splicing substrates with or without additional SR proteins. PCR analysis was done to quantify exon7 in these cells.
Key Results:
The most valuable result gathered from this experiment was the determined involvement of SRSF2 and SRSF3 in exon7 inclusion. Through knockdown of specific targets, an increased level of SMN protein was found to be present in these cells. This leads to further implications that these proteins could be therapeutic targets for Type I SMA patients.
Succeeding overexpression of SR proteins, the researchers observed a decrease in exon7 inclusion. Moreover, following successful knockdown of SR proteins in HeLa cells, it was found that exon7 inclusion increased significantly in 9 of 12 SR proteins. Specifically, exon7 inclusion was found to have the highest levels following SRSF3 knockdown. Following 40-50% knockdown of hnRNP A2B1 and U, a significant inclusion of exon7 resulted. This result confirmed that both SR and hnRNP proteins enhance splicing and through inhibiting their function, more exon 7 inclusion occurs.
Similar to the above experiments, the researchers decided to knockdown SR proteins in SMA cells and observe not only the effects on exon7 inclusion, but also SMN protein abundance. It was observed that upon knockdown of SRSF2 and 3, SMN protein levels increase. Thus, downregulation of these SR proteins leads to an increase in SMN protein levels. These results were consistent with the results obtained from HeLa cells. It was through this data that the researchers decided to examine the SRSF2 and 3 proteins more closely and deemed them to be the most effective therapeutic targets.
Broader Context:
There is a lower toxicity associated with targeting individual proteins such as splicing factors that have fundamental functions in the cell. Downregulation of single or multiple SR protein inhibitors of SMN2 exon7 inclusion could result in an improvement in full-length SMN protein. As a result, levels of SMN expression could increase to adequate therapeutic levels without fully disrupting other necessary functions of splicing factors.
Additional References:
MDA: Fighting Muscular Disease Website: mda.org/disease/spinal-muscular-atrophy/overview
Gene Cards: The Human Gene compendium Website: www.genecards.org