Heather Geist and Juliana Schneider
Summary of:
Wee, CD, Havens, MA, Jodelka, FM, Hastings, ML. (2014). Targeting SR Proteins Improves SMN Expression in Spinal Muscular Atrophy Cells. PLoS ONE, 9(12). e115205. doi:10.1371/journal.pone.0115205
Background:
What is SMA?
Spinal muscular atrophy (SMA) is an autosomal recessive genetic disease that is one of the most common inherited causes of pediatric mortality. SMA has occurs in 1 in 6000 live births and has a carrier frequency of 1 in 40. This disease affects the voluntary muscle movement controls of the nervous system. SMA is categorized as a degenerative neuromuscular disease, which involves the loss of motor neurons in the spinal cord. This in turn prevents the muscle from receiving signals, leading to muscular atrophy over time. The most common form of SMA is called chromosome 5 SMA, which has great variability in age of onset. For this reason it is typically classified into types 1 through 4. Our main focus will be on Type I SMA patients. Type I is correlated with the lowest level of functioning patients. Onset of symptoms in Type I patients usually occurs at birth or during infancy, leading to the greatest impact on motor function. The most drastically affected muscles are typically those closest to the center of the body and because of early onset, these individuals have high mortality rates.
What is SMA Caused by?
SMA is caused by deletions or mutations in the Survival of Motor Neuron I gene (SMN1) located on chromosome 5. SMN proteins are essential for conventional motor neuron function. If a deletion or mutation of SMN1 occurs, an unstable form of the SMN protein results. The SMN2 gene, also located on chromosome 5, has the ability to produce SMN protein at smaller volumes (Figure 1). The main aberration between SMN1 and 2 is the single nucleotide in exon7, which is thought to be an exon splice enhancer. As a result of this single nucleotide difference, SMN2 cannot compensate for the loss of SMN1. The SMN1 gene is telomeric. It contains more than 4 genes, so it is susceptible to rearrangements and deletions. SMN2, however, is centromeric and is not as prone to rearrangement. Interestingly, increasing the inclusion of exon7 escalates the abundance of SMN protein. The SMN2 gene, also located on chromosome 5, produces SMN protein at smaller volumes. Inclusion of exon7 has been shown to have efficacy in animal models of SMA and early human clinical trials.
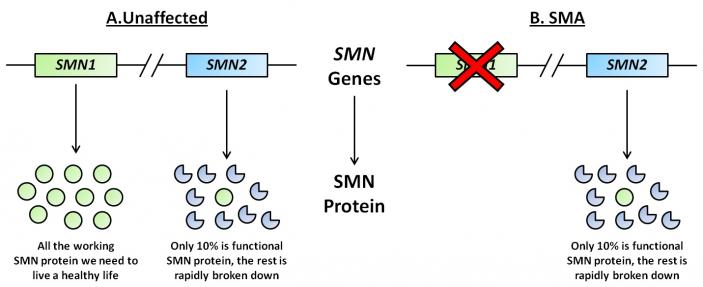 Figure 1. Representative diagram of SMN1 and SMN2 genes and subsequent proteins in unaffected and SMA individuals. (Image modified from http://www.smasupportuk.org.uk/blog/ research/azzouz-laboratory-gene-therapy-for-sma).
Figure 1. Representative diagram of SMN1 and SMN2 genes and subsequent proteins in unaffected and SMA individuals. (Image modified from http://www.smasupportuk.org.uk/blog/ research/azzouz-laboratory-gene-therapy-for-sma).
Splicing and its role in SMA:
Exons and introns undergo a period of pre-mRNA splicing. This is conducted by the spliceosome complex, which is comprised of 5 snRNPs and other splicing proteins/factors (Figure 2). The splicesome complex works to identify exons and introns through binding to specific consensus splicing sequences on both the 5’ and 3’ ends of mRNA. Multiple proteins and sequence elements have been shown to play a role in regulating the splicing of SMN exon7. Specifically, SRSF1, SRSF2, and SRSF9 are known to influence exon7 inclusion, but few other members of the SR protein family have been explored regarding their role in SMN2 exon7 splicing. In addition to the SR protein family, the hnRNP proteins have also been implicated in play a role in exon 7 inclusion or exclusion.
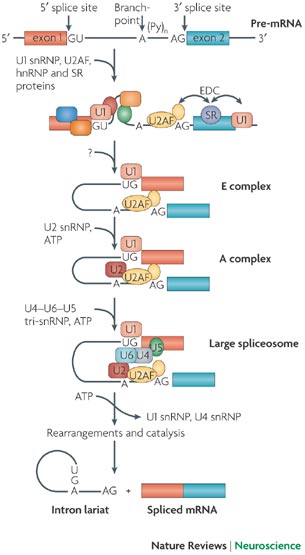 Figure 2. The spliceosome complex and process of pre-mRNA processing. (Image modified from http://www.nature.com/nrn/journal/v8/n11/box/nrn2237_BX1.html)
Figure 2. The spliceosome complex and process of pre-mRNA processing. (Image modified from http://www.nature.com/nrn/journal/v8/n11/box/nrn2237_BX1.html)
Question:
What characteristics and roles do SR and hnRNP proteins play in SMA disease pathogenesis? Do any of these proteins have therapeutic implications?
Experimental Approach:
Two main cell-lines were utilized throughout the experiments. The first was HeLa cells, which are derived from a cervical cancer tumor. The cells were treated and transfected with siRNA and Lipofectamine to knockdown specific SR and hnRNPs. In order to overexpress these proteins, expression vectors were transfected into the cells. The second cell-line used were human fibroblast cells derived from a Type I SMA patient only having one copy of SMN2. These cells were transfected as the HeLa cells were to knockdown the designated SR and hnRNP proteins. After cells were treated, RNA was isolated, and RT-PCR was conducted.
Proteins were extracted from the cells and immunoblots were performed by probing with 12 different antibodies directed at specific proteins. After quantitative analysis of fluorescence was performed, different levels of exon7 expression were observed. In vitro transcription was conducted on RNA splicing substrates with or without additional SR proteins. PCR analysis was done to quantify exon7 in these cells.
Key Results:
The most valuable result gathered from this experiment was the determined involvement of SRSF2 and SRSF3 in exon7 inclusion. Through knockdown of specific targets, an increased level of SMN protein was found to be present in these cells. This leads to further implications that these proteins could be therapeutic targets for Type I SMA patients.
Succeeding overexpression of SR proteins, the researchers observed a decrease in exon7 inclusion. Moreover, following successful knockdown of SR proteins in HeLa cells, it was found that exon7 inclusion increased significantly in 9 of 12 SR proteins. Specifically, exon7 inclusion was found to have the highest levels following SRSF3 knockdown. Following 40-50% knockdown of hnRNP A2B1 and U, a significant inclusion of exon7 resulted. This result confirmed that both SR and hnRNP proteins enhance splicing and through inhibiting their function, more exon 7 inclusion occurs.
Similar to the above experiments, the researchers decided to knockdown SR proteins in SMA cells and observe not only the effects on exon7 inclusion, but also SMN protein abundance. It was observed that upon knockdown of SRSF2 and 3, SMN protein levels increase. Thus, downregulation of these SR proteins leads to an increase in SMN protein levels. These results were consistent with the results obtained from HeLa cells. It was through this data that the researchers decided to examine the SRSF2 and 3 proteins more closely and deemed them to be the most effective therapeutic targets.
Broader Context:
There is a lower toxicity associated with targeting individual proteins such as splicing factors that have fundamental functions in the cell. Downregulation of single or multiple SR protein inhibitors of SMN2 exon7 inclusion could result in an improvement in full-length SMN protein. As a result, levels of SMN expression could increase to adequate therapeutic levels without fully disrupting other necessary functions of splicing factors.
Additional References:
MDA: Fighting Muscular Disease Website: mda.org/disease/spinal-muscular-atrophy/overview
Gene Cards: The Human Gene compendium Website: www.genecards.org
I find this article very interesting, and it seems like a crucial preliminary step in understanding Spinal Muscular Atrophy. One question that I have is how they chose to isolate the regulatory sequences around Exon 7. I am interested if other sequences would show the same results, and understanding more about how they chose to isolate this section.
The authors seem fairly confident in the potential therapeutic effects that SR knockdown, specifically that it would only take “modest down-regulation” for therapeutic efficacy. Their results show, in one case at least, about a 50% increase in SMN protein levels. Is there strong evidence that this amount of increase is sufficient to act therapeutically? Is their indication from previous research that this level of SMN protein would restore normal motor neuron function?
As the authors noted in the second part of the results, overexpression of some SR proteins (e.g. SRSF4, 6, and 9) did not have a significant effect on exon 7 splicing. They rationalize these results by saying 1) they may not be involved in exon 7 splicing, 2) there was no significant change because their levels of overexpression were insufficient, or 3) endogenous proteins are already optimized for regulating splicing and cannot be further enhanced. What else could the authors do to verify these results and justify why there was no significant effect with these SR proteins?
The authors chose to analyze the sequences flanking exon 7 within 50 nucleotides but acknowledge that there are antisense oligonucleotides directed to sequences more distant from exon 7 which have been identified to improve exon 7 inclusion. I was wondering why they chose to only look at the flanking sequences. Is there any benefit in one over the other, or was it just a need to limit their analyses and was chosen arbitrarily? On that note I’m curious to see if there have been studies looking at those more distant or if the authors plan to analyze those options in the future as well.
In this study, the authors conclude that the majority of SR proteins (SRF1 and more) and two hnRNPS are regulators of SMN2 exon 7 inclusion. How is it that so many proteins are responsible for the exon 7 inclusion? Also,the study identified regulators of exon 7 inclusion and also confirmed the activity of proteins that have been tested for exon 7 inclusion in recent studies. However, SRF9 had no significant affect on SRF9. It would be interesting to verify the role of SRF9 and see why it may not affect exon 7 inclusion.
I am very intrigued by the research that the authors in this article present. There are not many therapeutics available for spinal muscular atrophy, and this disease often effects infants so an effective therapeutic has been sought out for awhile. Although the authors determined that the SRSF2 and 3 proteins increase SMN levels, I’m curious if they investigated other areas on exon 7 that contribute to protein production. I’m also curious to see how they implement these findings into a therapeutic for this disease.
I found this article very interesting, but I am still unclear as to how the authors propose their findings be translated into therapies for SMA. They state that downregulation of single or multiple SR protein inhibitors of SMN2 exon 7 inclusion would be most effective, but how would they determine which inhibitors to use? Is it enough to just inhibit SRSF 2 and 3 or would inhibition of most of the proteins involved in the complex be necessary and how would that distinction be made?
I found the aim of this paper very interesting. While we all have learned that pre-mRNA can be alternatively spliced based on tissue location or specific promoters present in a certain cell type, I had not considered alternative slicing as a therapeutic option for disease. Here, the idea is clear, two types of the gene exist, and one (SMN2) could potentially act as the main gene (SMN1) if it expressed exon 7. I would be interested to see this effects in an animal model. In addition I am very curious as to how this type of therapy could be applied to other human disease where a redundant gene is not present.
I think it is interesting that the splicing proteins are interacting in such a complicated way. As I believe the authors mentioned, wouldn’t knocking down one of the genes have an effect that is based at least partly on how that protein interacts with the other components of the spliceosome? With such complicated interactions, how much does this tell you about the function of one protein? Additionally, I wonder if the spliceosome operates the same way in the cell-free condition?
This paper provides some interesting results to consider, but I think the claims of therapeutic benefit are weak based on the data presented in this paper. The effects of altering single splicing proteins are modest at best and I wonder if these subtle differences would lead to any therapeutic benefit. I also have concern that the method they used to measure the transcript levels (semi-quantitative RT-pcr) was not the best way to do this. We can discuss semi-quantitative pcr in class on Friday, but it is not as quantitative (hence the word “semi”!) as the RT-pcr methods we discussed/are using in our lab.
I could understand the basic mechanism of SMA! I think figure 4 is strongest figure that can support the main idea of the paper that depleting inhibitor (SRSF2/SRSF3) would increase SMN2 exon 7 inclusion and SMN protein that can lead to an therapeutic agent. Thus, I really like the fact that this paper has clear outline, which tells splicing factors are important. However, I agree that this needs more detail on therapies of SMA. This only identified the fact that knocking down SRSF3 could increase in SMN protein, which is related to SMA. Instead of identifying the fact, I think it should go further from exon 7 inclusion. Exon 7 inclusion wouldn’t be the only reason that lead to SMA. Even in figure 5, I think it should go further with molecular pathway of SMA instead of confirming knocking down SRSF3 could be a potential therapeutic target. I was wondering why they were using semi-quantitative rt-pcr. I also was wondering why the data they got about SRSF9 was different than previously published data. The reason they stated was ambiguous (vague).
After reading both the journal article and the blog post I am still finding it difficult to understand the purpose and function of the different cell lines that were used and transfected with in the experimental approach? I also would like to further elaborate on this idea of two genes coding for same protein with the exception that SMN2 cannot account for loss of SMN1 but it increases the relative protein levels upon inclusion of this exon 7. What is it biochemically that allows for different functions of SMN1 and 2 based on the presence of Exon 7?