Summary of:
Low, S. H., Vasanth, S., Larson, C. H., Mukherjee, S., Sharma, N., Kinter, M. T., Kane, M. E., Obara, T. & Weimbs, T. (2006). Polycystin-1, STAT6, and P100 Function in a Pathway that Transduces Ciliary Mechanosensation and Is Activated in Polycystic Kidney Disease
By: Lizz Reese and JT Stoner
What is ADPKD?
Autosomal dominant polycystic kidney disease (ADPKD) is one of the most common forms of polycystic kidney disease. It is known to occur in individuals and families of all different races and is estimated to currently impact the lives of 400,000+ people in the US. ADPKD usually presents in adulthood with about 50% of patients developing end-stage kidney disease by the age of 60.
This disease is characterized by fluid-filled cyst growth in both kidneys, which begin to replace much of the normal kidney mass. As a result, reduced function and organ failure may occur, requiring the patient to undergo dialysis and/or kidney transplant. PKD can also cause cysts in the liver as well as additional problems in the heart and blood vessels of the brain.
While there is no definitive treatment for ADPKD, treatments have been tailored to mitigate non-kidney symptoms (e.g. blood pressure) and pain (e.g. treated with painkillers and antidepressants). Kidney-related treatments typically aim to control buildup of acid and to prevent elevated phosphate levels.
What causes ADPKD?
ADPKD is genetically inherited in an autosomal dominant fashion. The mutated gene (either PKD1 or PKD2) causes renal cells to proliferate abnormally, resulting in the formation of fluid-filled cysts which eventually replace most of the normal renal tissue and lead to renal failure.
What is known?
Mutations in PKD1 or PKD2 are recognized as the underlying causes of ADPKD, with PKD1 being mutated in 85% of the cases. The function of the gene product, PC1, is poorly understood. PC1 is a large, integral membrane protein which is believed to have extracytoplasmic ligand binding domains, although ligands have yet to be identified. The C-terminal tail has been implicated in signal transduction pathways such as the wnt pathway, a pathway leading to AP-1 transcription factor activation, G protein signaling, calcium signaling, and activation of STAT1. It is unknown, however, if any of these pathways are altered in ADPKD.
In a process that is thought to incorporate PC1, primary cilia of renal epithelial cells act as mechanosensors that respond to changes in lumenal fluid and flow (see figure below). Moreover, it has been shown in a number of case that defects in cilia proteins can lead to renal cystic diseases in humans and animals, though PC1’s role remains unclear.
 V. Singla et al., Science (2006) Published by AAAS.
V. Singla et al., Science (2006) Published by AAAS.
Article Summary
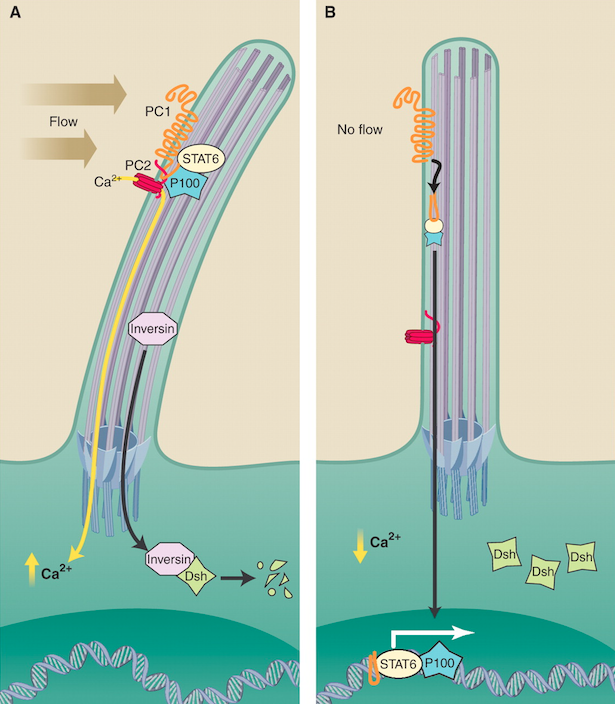
Autosomal dominant polycystic kidney disease (ADPKD) results from polycystin-1 (PC1) defects, which are poorly understood but known to implicate primary cilia. This paper identifies a novel mechanism of cilia function that leads to changes in gene expression via PC1 and shows that this pathway is inappropriately activated in ADPKD. Under normal conditions, the PC1 cytoplasmic tail interacts with transcription factor STAT6 and the coactivator P100 to stimulate STAT6-dependent gene expression. Termination of apical fluid flow results in nuclear translocation of STAT6. Under ADPKD conditions cyst-lining cells exhibit higher levels of nuclear STAT6, P100, and the PC1 tail. Exogenous expression of human PC1 in zebrafish embryos results in cyst formation.
Hypothesis
The signaling pathway that transduces a mechanical signal from primary cilia to changes in gene expression is inappropriately activated in ADPKD by the proteolytic cleavage and nuclear translocation of polycystin-1 (PC1).
Methods & Models
A number of different cells lines and models were used throughout the experiment: MDCK renal epithelial cell line (derived from a canine), COS-7 cells (derived from monkey kidney tissue), and HEK293T cells (human embryonic kidney cells) as well as normal and diseased human and mouse kidneys and zebrafish embryos. The MDCK and COS-7 cells were transfected with FLS-PC1 or CTM-PC1 to demonstrate that the cytoplasmic tail of PC1 localizes to the nucleus and to show that the C-terminal half of the PC1 tail is cleaved, released from the membrane, and targeted to the nucleus. Results were viewed via Western blots and immunostaining. Additionally, MDCK cells were utilized to demonstrate that the PC1 tail interacts with P100 via CTM-PC1 or NTM-PC1 transfection, Coomassie staining, and confocal/immunofluorescence microscopy. MDKC and HEK293T cells were used in experiments to show that the C-terminal half of PC1 tail interacts with STAT6 and activates STAT6-dependent transcription by transfection with FLS-PC1 or luciferase reporter constructs, respectively, followed by Western blotting and reporter assays. MDCK cells continued to be used for experimentation to determine that STAT6 localized to cilia and translocated to the nucleus under ‘no-flow’ conditions via immunofluorescence microscopy with tagged STAT6. Immunohistochemistry was used to detect STAT6 in human ADPKD and normal kidneys. Primary cilia were detected with H&E staining. Zebrafish embryos were utilized to show that the human PC1 tail causes pronephric cysts by injecting one population with FLS-PC1 and monitoring human PC1 mRNA via RT-PCR at 3 days post fertilization.
Key Findings
Because the function of PC1 is poorly understood, these authors aimed to explore and better understand how its binding to transcription factor STAT6 and coactivator P100 affects the relationship between ciliary mechanosensation and the onset of autosomal dominant polycystic kidney disease. The most significant discoveries in this study are outlined below:
- Full length PC1 is vulnerable to rapid proteasomal degradation, localizes to the nucleoplasm, and is overexpressed in ADPKD conditions.
- In a diseased kidney, the C-terminal half of the PC1 tail is cleaved not only at a GPS domain, but another site as well, which leads to its release from the membrane.
- Different constructs of PC1 localize to either the cytoplasm (NTS-PC1) or the nucleus (CTS-PC1 and CTSP-PC1), the latter of which are mediated by the C-terminal half of the PC1 tail, suggesting that the tail undergoes nuclear shuttling.
- P100, which is a coactivator to transcription factor STAT6, binds to the PC1 tail. It localizes to the basal body and primary cilia in polarized MDCK cells and is overexpressed in ADPKD renal tissue, specifically in cyst-lining epithelial cells.
- In addition to PC1 binding with P100, it also binds with transcription factor STAT6 to stimulate STAT6-dependent transcription. More specifically, the C-terminal half of the cytoplasmic tail positively regulates STAT6-dependent transcription.
- STAT6 was found to be moderately expressed in renal epithelial cells of normal human kidneys, overexpressed in cyst-lining epithelial cells of ADPKD kidneys, and is involved in transduction of mechanical signal originating at primary cilia. It is activated under no-flow conditions, translocating from primary cilia to nuclei of renal epithelial cells. Further experimentation suggests that STAT6-dependent gene expression is highly upregulated in ADPKD.
- Using a zebrafish model, the authors were able to demonstrate that exogenous overexpression of soluble PC1 tail alone can stimulate renal cyst formation.
Broader Context
The primary findings outlined above have allowed the authors to construct the following signaling mechanism, which demonstrates how PC1 modulates STAT6-dependent transcription. Upon initial cleavage of the cytoplasmic tail of PC1, the membrane-bound C-terminal half of the tail is freed from the membrane. It can then bind to STAT6 and the transcriptional coactivator P100 and translocate into the nucleus where it induces STAT6-dependent transcription. Evidence demonstrating that, in the absence of apical fluid flow, STAT6 translocates from primary cilia to the nucleus allowed the authors to hypothesize that the PC1/STAT6/P100 pathway may have greater implications in the relationship between mechanical signal transduction and transcriptional response. Moreover, because this pathway is highly upregulated in human ADPKD cysts, it is thought to have a significant role in the progression of this human disease. With a better understanding of how PC1/STAT6/P100 pathway is regulated, researchers can now focus their attention on specific components of this pathway that may serve as therapeutic targets for the treatment of ADPKD.
Links
ADPKD: https://www.genome.gov/20019622
Polycystins and mechanosensation: http://onlinelibrary.wiley.com/doi/10.1002/bies.20069/epdf
One of the things I found most interesting about this research paper are the varied models the researchers used. For the authors’ figures, I especially like Figure 7. Although I do not believe it is the strongest figure, the morphology changes and the histology is fascinating. I believe that Figure 5 is their strongest data figure, as it is one of the few figures in this paper that quantifies their data well. Overall, I am excited to learn more about this topic, and to better understand PC1.
The researchers use a variety of cell lines to conduct these experiments, which shows that this phenotype is conserved in the disease throughout many species. The results that are shown provide a lot of information, but I found it to be more observational than quantitative in some cases and in some cases further quantification of observations may have made the results stronger in the figures. Another component I found interesting was the role of cilia in ADPKD. I typically think of cilia in terms of digestion and its role in that process. I was interested to find it plays an important role in PKD and is altered by PC1.
The article concludes that p100 is a coactivator of transcription factor STAT6 and that it may exist in a stable complex with PC1 which localizes to cilia. Furthermore, the article cites another article that says that p100 has been shown to increase in mammary epithelial cells during lactation. How does this information affect the conclusion of the study. Might we discuss how this information can affect the possible role of p100 in ADPKD. Could the use of more mammary epithelial cells during this study be useful in observing the effect of p100?
I found this article very interesting, but I still do not fully understand the interactions between PC1, STAT6, and P100. Because P100 is a coactivator or STAT6, would blocking its activity also block STAT6 activity? If so, would that make it a better target for therapies than PC1 or STAT6? Also is the over-expression of these factors all related to mutations in PKD1 and PKD2 or just PC1?
After an initial reading of the paper, I would appreciate help clearing up some confusion. What is their proposed explanation for how the mutation in PKD1 or PKD2 leads to the observed pathophysiology? It is my understanding that the upregulation of nuclear PC1 is due to the fact that it is cleaved and released from the membrane in ADPKD conditions. However, I do not understand how the mutation in PKD1 or PKD2 leads to this condition. Does it alter the protein in some way as to make it more likely to be cleaved?
I thought this was an interesting paper. I do not have much knowledge on the subject matter–so it was great to read about something new. However, to me it appeared as though they did not have very much convincing evidence. Particularly Figure 6 (a-h mainly) did not seem to show anything or contribute to the hypothesis. No figure stood out as particularly meaningful to me. I did think that it was interesting that they used zebrafish. I would like to know how the results of this study would contribute to an effective treatment of ADPKD.
Although PC1’s role in ADPKA is unclear, it is interesting that the defects in the cilia protein are known to contribute to renal real cystic diseases. Full length PC1 is over expressed in APKD conditions, and the mechanism from the experiments shown that PC1 modulates STAT6-dependent transcription. Due to the up regulation of the PC1/STAT6/P100 in the disease, I’m curious to see different experiments involving different components of the mechanism and what components of the mechanism that would be specific targets in the disease. This research is intriguing since there is not a lot of data or information about different therapeutics for ADPKA.
While the exact mechanisms of PC1 is not known, I thought the association of a protein mutation and a mechanical change in altered cilia function was really interesting and more clear here than in many other cases. I would be really interested to see if this same concept translates to other areas with cilia in the body, which is obviously a large number of locations. Perhaps a better understanding for this association will help make discoveries in polycystic kidney disease along with many other disorders involving cilia mechanosensation.
I thought one of the coolest parts of this paper was the variety of PC1 constructs used. Although we have seen many different molecular techniques this semester, this is the first time we have seen the use of modified versions of a normal protein, with sections removed or tags added, etc. I thought that the way this was used to derive the localizing segments of PC1 was very interesting! Additionally, I was a fan of figure 6. To me, showing cells with and without shaking was a great visual for how flow could change the localization patterns of STAT6.
I liked that they used zebrafish to demonstrate a key role of the PC1 tail in cyst formation, but this experiment was not an in vivo model for the role of cilia in PC1 signaling, which was a major focus of the paper. Are there any animal models where PC1 mutation in combination with ciliary localization signaling could be studied? Also this paper was published in 2006…I wonder what has been done since to confirm/advance these findings??
In this paper, it was interesting that they put model for the role of PC1 with P100 and STAT6 in ADPKD. I am wondering more information about PC1/STAT6/P100 pathway. What would be the reason where their data contrast with other primary literature? They observed AP-1 stimulation with a membrane-anchored fusion protein of the PC1 tail unlike other research group didn’t.
P100’s role in the interaction with PC1 and localization in the primary cilia of cells with ADPKD phenotype. Understanding how different protein complexes function in cilia is a research area that I have not given much thought of before. How ever, cilia and its dysfunction plays a role in many different human diseases. Knowing exactly how the dysregulation of proteins involved in the function of cilia gives new avenues for research and therapeutics.
Given that the C-terminus tail of the PC1 is what allows for activation of STAT6, I would like some clarification of how the causes of this disease are categorized, specifically are all 85% of the patients with mutations in PKD1 going to have this exact mechanism occurring? And does the mutation in PC1 rely on its interaction with STAT6 to have pathogenic effect that is observed in this article?